LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_506.5wLII_11364_47
Total number of 3D structures: 71
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
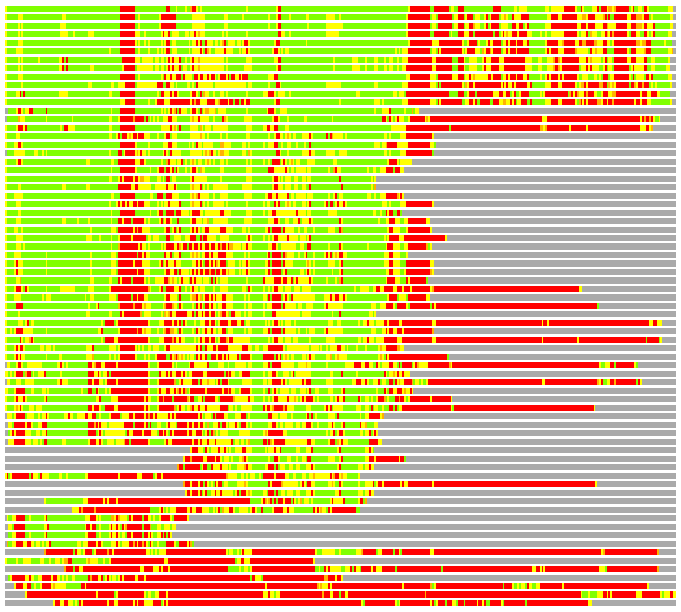
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 2it1_A |
362 |
361 |
299 |
1.36 |
16.72 |
79.387 |
20.522 |
T P |
| 1vci_A |
353 |
361 |
275 |
1.89 |
14.91 |
67.199 |
13.787 |
T P |
| 1v43_A |
353 |
361 |
272 |
1.89 |
14.71 |
66.380 |
13.687 |
T P |
| 2yyz_A |
358 |
361 |
278 |
1.96 |
14.75 |
65.553 |
13.505 |
T P |
| 2awn_B |
374 |
361 |
265 |
1.91 |
14.34 |
65.026 |
13.210 |
T P |
| 1q1b_A |
367 |
361 |
273 |
1.98 |
15.38 |
62.256 |
13.138 |
T P |
| 1oxx_K |
352 |
361 |
277 |
2.06 |
9.75 |
61.221 |
12.802 |
T P |
| 1oxs_C |
352 |
361 |
277 |
2.09 |
10.11 |
61.218 |
12.632 |
T P |
| 2r6g_B |
372 |
361 |
261 |
1.83 |
14.56 |
60.193 |
13.526 |
T P |
| 2d62_A |
375 |
361 |
262 |
2.08 |
13.36 |
59.962 |
11.996 |
T P |
| 1z47_A |
345 |
361 |
250 |
1.90 |
12.80 |
58.876 |
12.506 |
T P |
| 1g29_1 |
372 |
361 |
239 |
1.92 |
14.23 |
56.651 |
11.806 |
T P |
| 2onk_A |
240 |
361 |
198 |
1.76 |
13.13 |
51.031 |
10.652 |
T P |
| 3dhw_C |
343 |
361 |
207 |
1.92 |
12.08 |
50.945 |
10.233 |
T P |
| 3d31_A |
348 |
361 |
208 |
1.96 |
13.94 |
50.890 |
10.112 |
T P |
| 2olj_A |
242 |
361 |
201 |
1.86 |
11.44 |
49.862 |
10.249 |
T P |
| 1ji0_A |
240 |
361 |
199 |
1.81 |
15.08 |
49.606 |
10.426 |
T P |
| 1b0u_A |
258 |
361 |
202 |
1.97 |
10.40 |
48.321 |
9.735 |
T P |
| 2yz2_A |
266 |
361 |
195 |
1.92 |
14.87 |
48.165 |
9.674 |
T P |
| 1g6h_A |
254 |
361 |
186 |
1.71 |
11.83 |
47.311 |
10.251 |
T P |
| 1f3o_A |
232 |
361 |
186 |
1.70 |
12.37 |
46.906 |
10.336 |
T P |
| 2pcj_A |
223 |
361 |
184 |
1.83 |
13.04 |
45.971 |
9.553 |
T P |
| 1g9x_A |
253 |
361 |
188 |
1.94 |
11.17 |
45.121 |
9.210 |
T P |
| 3c41_J |
242 |
361 |
197 |
1.88 |
12.69 |
41.996 |
9.954 |
T P |
| 1gaj_A |
253 |
361 |
183 |
1.89 |
10.93 |
41.113 |
9.191 |
T P |
| 1mt0_A |
241 |
361 |
193 |
1.98 |
13.99 |
40.033 |
9.260 |
T P |
| 2ixe_A |
251 |
361 |
186 |
1.88 |
15.05 |
39.641 |
9.378 |
T P |
| 1vpl_A |
238 |
361 |
185 |
1.88 |
16.22 |
39.543 |
9.332 |
T P |
| 2hyd_A |
578 |
361 |
180 |
2.00 |
13.89 |
38.279 |
8.582 |
T P |
| 1jj7_A |
251 |
361 |
180 |
1.97 |
14.44 |
37.944 |
8.699 |
T P |
| 2ixf_A |
255 |
361 |
182 |
1.99 |
13.74 |
37.658 |
8.702 |
T P |
| 2ixg_A |
254 |
361 |
175 |
1.88 |
13.14 |
37.604 |
8.835 |
T P |
| 2ff7_A |
243 |
361 |
184 |
1.95 |
11.96 |
37.519 |
8.956 |
T P |
| 2nq2_C |
251 |
361 |
176 |
2.06 |
12.50 |
36.613 |
8.130 |
T P |
| 3b60_A |
572 |
361 |
181 |
2.07 |
16.57 |
36.374 |
8.360 |
T P |
| 2qi9_C |
248 |
361 |
178 |
2.04 |
15.17 |
36.071 |
8.329 |
T P |
| 1l2t_B |
232 |
361 |
169 |
1.98 |
11.83 |
35.610 |
8.138 |
T P |
| 1yqt_A |
515 |
361 |
170 |
2.00 |
14.71 |
35.379 |
8.078 |
T P |
| 1l7v_C |
231 |
361 |
178 |
2.13 |
14.61 |
35.182 |
7.985 |
T P |
| 3bk7_A |
593 |
361 |
172 |
2.06 |
16.28 |
34.527 |
7.965 |
T P |
| 2zu0_C |
247 |
361 |
172 |
2.34 |
12.21 |
33.488 |
7.048 |
T P |
| 2d3w_A |
244 |
361 |
165 |
2.14 |
15.15 |
32.901 |
7.364 |
T P |
| 2iw3_B |
980 |
361 |
176 |
2.38 |
10.23 |
32.262 |
7.110 |
T P |
| 2vf7_A |
815 |
361 |
151 |
2.06 |
9.93 |
31.070 |
6.982 |
T P |
| 2ix3_A |
972 |
361 |
172 |
2.36 |
8.72 |
31.051 |
6.999 |
T P |
| 2r6f_A |
899 |
361 |
151 |
2.13 |
12.58 |
30.658 |
6.763 |
T P |
| 2pjz_A |
261 |
361 |
154 |
2.17 |
13.64 |
30.500 |
6.772 |
T P |
| 2ix8_A |
975 |
361 |
160 |
2.33 |
11.88 |
29.327 |
6.579 |
T P |
| 1e69_A |
263 |
361 |
142 |
2.25 |
9.86 |
27.754 |
6.040 |
T P |
| 1w1w_B |
327 |
361 |
144 |
2.19 |
9.03 |
27.724 |
6.287 |
T P |
| 3euj_A |
437 |
361 |
146 |
2.28 |
11.64 |
27.409 |
6.125 |
T P |
| 2o5v_A |
358 |
361 |
140 |
2.35 |
11.43 |
26.056 |
5.724 |
T P |
| 1f2u_B |
145 |
361 |
86 |
2.06 |
6.98 |
19.299 |
3.988 |
T P |
| 1xew_Y |
160 |
361 |
82 |
2.11 |
3.66 |
19.157 |
3.713 |
T P |
| 1ii8_B |
174 |
361 |
84 |
2.06 |
7.14 |
18.952 |
3.882 |
T P |
| 1qvr_A |
803 |
361 |
98 |
2.48 |
14.29 |
17.535 |
3.806 |
T P |
| 1xex_B |
161 |
361 |
88 |
2.23 |
5.68 |
17.372 |
3.784 |
T P |
| 1us8_B |
140 |
361 |
82 |
2.13 |
8.54 |
17.162 |
3.678 |
T P |
| 2ax4_A |
198 |
361 |
76 |
2.16 |
17.11 |
15.729 |
3.357 |
T P |
| 1jsq_A |
450 |
361 |
80 |
2.42 |
8.75 |
14.549 |
3.177 |
T P |
| 1ii8_A |
195 |
361 |
68 |
2.22 |
13.24 |
14.412 |
2.934 |
T P |
| 1f2u_A |
149 |
361 |
65 |
2.11 |
13.85 |
14.244 |
2.937 |
T P |
| 1us8_A |
129 |
361 |
63 |
2.01 |
14.29 |
14.037 |
2.986 |
T P |
| 1xew_X |
147 |
361 |
71 |
2.39 |
18.31 |
13.562 |
2.856 |
T P |
| 1z5v_A |
412 |
361 |
71 |
2.54 |
7.04 |
13.200 |
2.693 |
T P |
| 1pf4_A |
520 |
361 |
64 |
2.32 |
14.06 |
12.878 |
2.640 |
T P |
| 3cb2_A |
432 |
361 |
60 |
2.63 |
11.67 |
11.497 |
2.201 |
T P |
| 1qhl_A |
203 |
361 |
52 |
2.60 |
17.31 |
9.839 |
1.923 |
T P |
| 1t9h_A |
287 |
361 |
55 |
2.71 |
3.64 |
9.640 |
1.960 |
T P |
| 2uuu_A |
550 |
361 |
51 |
2.78 |
7.84 |
9.611 |
1.770 |
T P |
| 2i1j_A |
481 |
361 |
45 |
2.68 |
4.44 |
8.700 |
1.618 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]