LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_509.5wLII_11364_59
Total number of 3D structures: 140
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
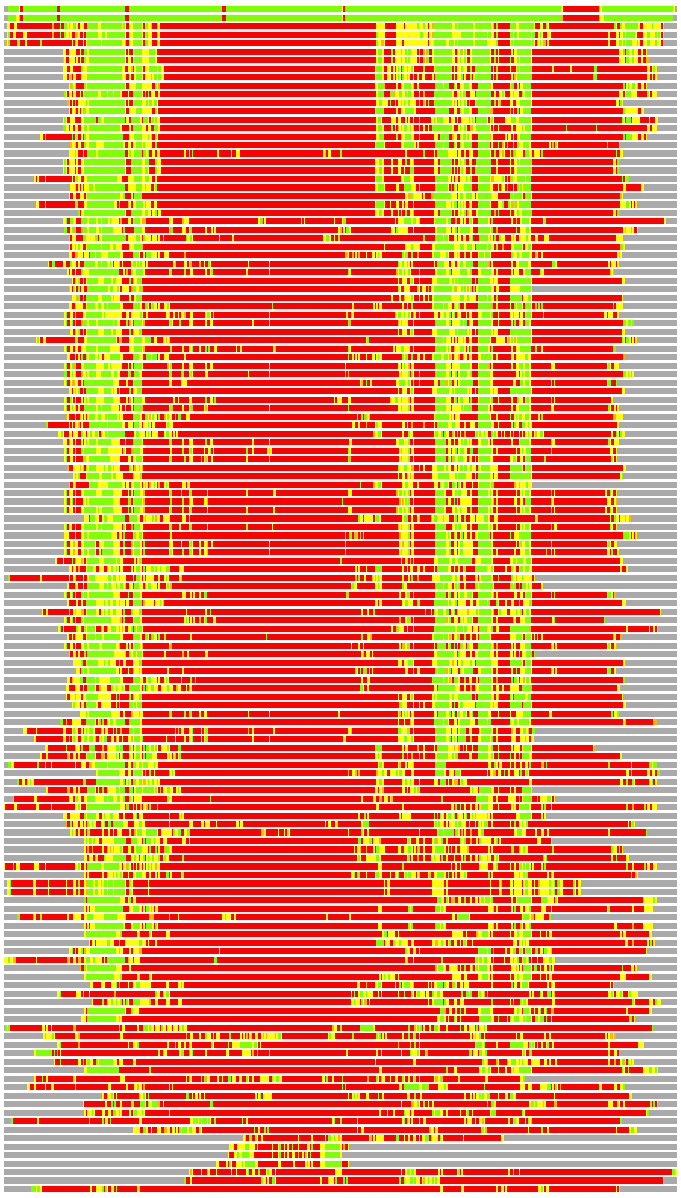
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1e9r_B |
422 |
444 |
405 |
0.61 |
30.37 |
90.639 |
56.995 |
T P |
| 1gl6_B |
420 |
444 |
404 |
0.78 |
30.45 |
89.984 |
45.863 |
T P |
| 2iut_A |
408 |
444 |
179 |
2.11 |
18.99 |
28.705 |
8.097 |
T P |
| 2iuu_A |
408 |
444 |
180 |
2.23 |
19.44 |
28.005 |
7.739 |
T P |
| 2ius_D |
393 |
444 |
179 |
2.24 |
17.88 |
27.968 |
7.665 |
T P |
| 1pv4_A |
408 |
444 |
144 |
2.19 |
8.33 |
22.531 |
6.293 |
T P |
| 1pvo_A |
408 |
444 |
146 |
2.21 |
8.90 |
22.527 |
6.319 |
T P |
| 2hld_D |
470 |
444 |
133 |
2.11 |
13.53 |
21.612 |
6.016 |
T P |
| 3fks_F |
471 |
444 |
133 |
2.21 |
15.04 |
21.320 |
5.759 |
T P |
| 2eww_A |
343 |
444 |
121 |
2.06 |
14.88 |
20.553 |
5.603 |
T P |
| 3b2q_A |
436 |
444 |
136 |
2.23 |
5.88 |
20.247 |
5.827 |
T P |
| 2ht1_A |
324 |
444 |
133 |
2.27 |
10.53 |
20.175 |
5.621 |
T P |
| 2ewv_A |
343 |
444 |
120 |
2.06 |
14.17 |
20.134 |
5.556 |
T P |
| 1sky_E |
470 |
444 |
133 |
2.35 |
14.29 |
19.800 |
5.438 |
T P |
| 2dpy_B |
426 |
444 |
127 |
2.19 |
10.24 |
19.543 |
5.534 |
T P |
| 1p9r_A |
378 |
444 |
121 |
2.08 |
13.22 |
19.465 |
5.542 |
T P |
| 2gsz_A |
343 |
444 |
115 |
2.10 |
14.78 |
19.263 |
5.234 |
T P |
| 2is6_A |
654 |
444 |
119 |
2.29 |
11.76 |
19.194 |
4.989 |
T P |
| 1tf7_A |
484 |
444 |
115 |
2.10 |
14.78 |
19.167 |
5.219 |
T P |
| 2gbl_A |
506 |
444 |
114 |
2.05 |
13.16 |
18.979 |
5.311 |
T P |
| 2gza_C |
336 |
444 |
123 |
2.28 |
14.63 |
18.978 |
5.163 |
T P |
| 2eyu_A |
247 |
444 |
118 |
2.16 |
16.10 |
18.779 |
5.219 |
T P |
| 1sxj_C |
322 |
444 |
108 |
2.10 |
17.59 |
18.399 |
4.904 |
T P |
| 1g6o_A |
323 |
444 |
116 |
2.26 |
14.66 |
18.358 |
4.918 |
T P |
| 2gbl_C |
488 |
444 |
111 |
2.12 |
16.22 |
18.320 |
5.009 |
T P |
| 2r6f_A |
899 |
444 |
116 |
2.38 |
15.52 |
17.919 |
4.685 |
T P |
| 1l2t_B |
232 |
444 |
118 |
2.34 |
10.17 |
17.886 |
4.838 |
T P |
| 1uaa_A |
636 |
444 |
119 |
2.61 |
13.45 |
17.753 |
4.389 |
T P |
| 1sxj_B |
316 |
444 |
110 |
2.24 |
15.45 |
17.539 |
4.705 |
T P |
| 1d2m_A |
552 |
444 |
115 |
2.50 |
14.78 |
17.435 |
4.427 |
T P |
| 2ix8_A |
975 |
444 |
116 |
2.42 |
11.21 |
17.363 |
4.602 |
T P |
| 1vpl_A |
238 |
444 |
112 |
2.33 |
12.50 |
17.181 |
4.616 |
T P |
| 1in8_A |
298 |
444 |
101 |
2.04 |
22.77 |
17.143 |
4.725 |
T P |
| 2qby_A |
366 |
444 |
111 |
2.31 |
18.02 |
17.140 |
4.602 |
T P |
| 1sxj_E |
317 |
444 |
103 |
2.11 |
16.50 |
17.101 |
4.659 |
T P |
| 2fdc_B |
585 |
444 |
109 |
2.16 |
14.68 |
17.078 |
4.812 |
T P |
| 2pjz_A |
261 |
444 |
124 |
2.66 |
10.48 |
17.065 |
4.497 |
T P |
| 2yz2_A |
266 |
444 |
113 |
2.29 |
14.16 |
17.045 |
4.721 |
T P |
| 1in7_A |
298 |
444 |
101 |
2.01 |
22.77 |
16.995 |
4.796 |
T P |
| 2pt7_A |
323 |
444 |
112 |
2.34 |
16.96 |
16.924 |
4.599 |
T P |
| 1r0z_C |
277 |
444 |
113 |
2.32 |
14.16 |
16.905 |
4.672 |
T P |
| 1c4o_A |
504 |
444 |
113 |
2.49 |
13.27 |
16.871 |
4.364 |
T P |
| 3c41_J |
242 |
444 |
114 |
2.41 |
14.04 |
16.856 |
4.543 |
T P |
| 2pcj_A |
223 |
444 |
110 |
2.26 |
15.45 |
16.831 |
4.662 |
T P |
| 2awn_B |
374 |
444 |
108 |
2.28 |
15.74 |
16.802 |
4.544 |
T P |
| 1j7k_A |
299 |
444 |
100 |
2.03 |
23.00 |
16.798 |
4.690 |
T P |
| 3b60_A |
572 |
444 |
112 |
2.48 |
12.50 |
16.766 |
4.349 |
T P |
| 2olj_A |
242 |
444 |
110 |
2.28 |
10.91 |
16.728 |
4.621 |
T P |
| 2d7d_A |
621 |
444 |
109 |
2.38 |
13.76 |
16.701 |
4.398 |
T P |
| 2pl3_A |
232 |
444 |
109 |
2.42 |
14.68 |
16.615 |
4.319 |
T P |
| 2gk6_A |
602 |
444 |
118 |
2.53 |
12.71 |
16.592 |
4.480 |
T P |
| 1b0u_A |
258 |
444 |
108 |
2.39 |
12.04 |
16.546 |
4.345 |
T P |
| 1mt0_A |
241 |
444 |
111 |
2.32 |
12.61 |
16.519 |
4.588 |
T P |
| 1g9x_A |
253 |
444 |
109 |
2.32 |
11.01 |
16.474 |
4.504 |
T P |
| 2chq_A |
313 |
444 |
107 |
2.44 |
14.02 |
16.443 |
4.221 |
T P |
| 1in4_A |
298 |
444 |
99 |
2.12 |
23.23 |
16.416 |
4.468 |
T P |
| 2va8_B |
695 |
444 |
107 |
2.38 |
17.76 |
16.364 |
4.322 |
T P |
| 2pmk_A |
243 |
444 |
108 |
2.37 |
9.26 |
16.327 |
4.365 |
T P |
| 2it1_A |
362 |
444 |
103 |
2.37 |
10.68 |
16.261 |
4.175 |
T P |
| 2ff7_A |
243 |
444 |
107 |
2.34 |
10.28 |
16.214 |
4.382 |
T P |
| 2ibm_A |
780 |
444 |
114 |
2.62 |
12.28 |
16.125 |
4.197 |
T P |
| 2ffb_A |
243 |
444 |
102 |
2.27 |
9.80 |
16.116 |
4.295 |
T P |
| 1g6h_A |
254 |
444 |
103 |
2.25 |
11.65 |
16.105 |
4.379 |
T P |
| 1oxs_C |
352 |
444 |
110 |
2.47 |
12.73 |
16.095 |
4.285 |
T P |
| 1oxx_K |
352 |
444 |
109 |
2.46 |
12.84 |
16.083 |
4.255 |
T P |
| 1r6b_X |
704 |
444 |
98 |
2.17 |
13.27 |
16.080 |
4.320 |
T P |
| 1wp9_A |
479 |
444 |
108 |
2.42 |
11.11 |
16.033 |
4.289 |
T P |
| 3dkp_A |
240 |
444 |
110 |
2.58 |
19.09 |
16.008 |
4.099 |
T P |
| 2db3_A |
420 |
444 |
105 |
2.27 |
12.38 |
15.993 |
4.436 |
T P |
| 1v43_A |
353 |
444 |
101 |
2.23 |
13.86 |
15.987 |
4.335 |
T P |
| 3b85_A |
187 |
444 |
109 |
2.49 |
17.43 |
15.898 |
4.208 |
T P |
| 2c9o_A |
398 |
444 |
107 |
2.50 |
10.28 |
15.854 |
4.112 |
T P |
| 1vci_A |
353 |
444 |
100 |
2.14 |
13.00 |
15.845 |
4.472 |
T P |
| 1qvr_A |
803 |
444 |
102 |
2.22 |
7.84 |
15.836 |
4.390 |
T P |
| 1t5l_A |
595 |
444 |
101 |
2.17 |
14.85 |
15.780 |
4.454 |
T P |
| 2yyz_A |
358 |
444 |
100 |
2.46 |
19.00 |
15.711 |
3.905 |
T P |
| 1fnn_A |
379 |
444 |
98 |
2.26 |
15.31 |
15.651 |
4.150 |
T P |
| 2chg_A |
223 |
444 |
102 |
2.34 |
16.67 |
15.577 |
4.181 |
T P |
| 1g29_1 |
372 |
444 |
101 |
2.28 |
9.90 |
15.558 |
4.246 |
T P |
| 1d9z_A |
590 |
444 |
101 |
2.22 |
15.84 |
15.518 |
4.345 |
T P |
| 2z0m_A |
331 |
444 |
109 |
2.48 |
13.76 |
15.510 |
4.221 |
T P |
| 1ixs_B |
315 |
444 |
97 |
2.21 |
23.71 |
15.435 |
4.195 |
T P |
| 1iqp_A |
326 |
444 |
95 |
2.29 |
16.84 |
15.433 |
3.969 |
T P |
| 1gaj_A |
253 |
444 |
105 |
2.39 |
13.33 |
15.429 |
4.214 |
T P |
| 1um8_A |
327 |
444 |
94 |
2.13 |
8.51 |
15.217 |
4.206 |
T P |
| 2iw3_B |
980 |
444 |
99 |
2.45 |
9.09 |
15.145 |
3.877 |
T P |
| 2ix3_A |
972 |
444 |
104 |
2.55 |
10.58 |
15.065 |
3.918 |
T P |
| 2i4i_A |
408 |
444 |
101 |
2.44 |
14.85 |
15.063 |
3.973 |
T P |
| 3eiq_D |
371 |
444 |
99 |
2.45 |
12.12 |
14.845 |
3.880 |
T P |
| 2j37_W |
479 |
444 |
94 |
2.33 |
11.70 |
14.845 |
3.867 |
T P |
| 1g20_H |
262 |
444 |
98 |
2.51 |
7.14 |
14.781 |
3.761 |
T P |
| 2px0_A |
258 |
444 |
92 |
2.39 |
10.87 |
14.399 |
3.695 |
T P |
| 1wrb_A |
232 |
444 |
95 |
2.38 |
5.26 |
14.092 |
3.833 |
T P |
| 1goj_A |
354 |
444 |
94 |
2.54 |
13.83 |
14.082 |
3.566 |
T P |
| 1fts_A |
295 |
444 |
89 |
2.51 |
8.99 |
13.965 |
3.416 |
T P |
| 3d31_A |
348 |
444 |
95 |
2.56 |
11.58 |
13.905 |
3.566 |
T P |
| 1e69_A |
263 |
444 |
101 |
2.70 |
11.88 |
13.879 |
3.613 |
T P |
| 1d9x_A |
590 |
444 |
94 |
2.57 |
10.64 |
13.797 |
3.521 |
T P |
| 1m6n_A |
802 |
444 |
97 |
2.60 |
11.34 |
13.747 |
3.587 |
T P |
| 1de0_A |
289 |
444 |
93 |
2.76 |
7.53 |
13.584 |
3.246 |
T P |
| 1tf5_A |
775 |
444 |
94 |
2.67 |
8.51 |
13.543 |
3.399 |
T P |
| 2qy9_A |
300 |
444 |
92 |
2.72 |
10.87 |
13.535 |
3.258 |
T P |
| 2afh_E |
289 |
444 |
92 |
2.57 |
10.87 |
13.508 |
3.446 |
T P |
| 1mys_A |
800 |
444 |
89 |
2.76 |
14.61 |
13.485 |
3.113 |
T P |
| 2mys_A |
800 |
444 |
89 |
2.76 |
13.48 |
13.485 |
3.113 |
T P |
| 2p67_A |
302 |
444 |
86 |
2.19 |
13.95 |
13.348 |
3.759 |
T P |
| 2hf9_A |
211 |
444 |
79 |
2.31 |
11.39 |
12.761 |
3.273 |
T P |
| 2otg_A |
800 |
444 |
79 |
2.55 |
12.66 |
12.664 |
2.976 |
T P |
| 2hf8_A |
211 |
444 |
76 |
2.35 |
11.84 |
12.435 |
3.099 |
T P |
| 1cke_A |
212 |
444 |
80 |
2.46 |
11.25 |
12.407 |
3.124 |
T P |
| 1yrb_B |
260 |
444 |
86 |
2.85 |
12.79 |
12.372 |
2.919 |
T P |
| 1rz3_A |
184 |
444 |
80 |
2.44 |
8.75 |
12.364 |
3.156 |
T P |
| 2owm_A |
328 |
444 |
87 |
2.62 |
11.49 |
12.356 |
3.197 |
T P |
| 2bdt_A |
172 |
444 |
78 |
2.37 |
14.10 |
12.259 |
3.153 |
T P |
| 2fem_A |
212 |
444 |
79 |
2.44 |
12.66 |
12.116 |
3.114 |
T P |
| 1xdb_A |
289 |
444 |
82 |
2.75 |
7.32 |
12.021 |
2.877 |
T P |
| 1ozq_A |
372 |
444 |
85 |
2.69 |
10.59 |
11.944 |
3.041 |
T P |
| 1rw4_A |
271 |
444 |
81 |
2.67 |
7.41 |
11.928 |
2.922 |
T P |
| 2ax4_A |
198 |
444 |
71 |
2.12 |
16.90 |
11.883 |
3.204 |
T P |
| 1kag_A |
158 |
444 |
74 |
2.12 |
13.51 |
11.838 |
3.335 |
T P |
| 3pgk_A |
415 |
444 |
78 |
2.63 |
8.97 |
11.829 |
2.855 |
T P |
| 2v3c_C |
403 |
444 |
81 |
2.88 |
6.17 |
11.426 |
2.722 |
T P |
| 1tpz_A |
395 |
444 |
75 |
2.53 |
6.67 |
11.221 |
2.850 |
T P |
| 2o2k_B |
333 |
444 |
69 |
2.58 |
5.80 |
11.105 |
2.575 |
T P |
| 1xzp_A |
456 |
444 |
72 |
2.68 |
1.39 |
10.659 |
2.591 |
T P |
| 2qpt_A |
476 |
444 |
66 |
2.05 |
15.15 |
10.588 |
3.074 |
T P |
| 3dm5_A |
416 |
444 |
72 |
2.78 |
5.56 |
10.520 |
2.497 |
T P |
| 1tq4_A |
396 |
444 |
70 |
2.66 |
15.71 |
10.368 |
2.538 |
T P |
| 1tqd_A |
385 |
444 |
65 |
2.80 |
3.08 |
9.300 |
2.245 |
T P |
| 1wf3_A |
296 |
444 |
58 |
2.61 |
1.72 |
8.700 |
2.139 |
T P |
| 1kdo_A |
223 |
444 |
55 |
2.52 |
5.45 |
8.372 |
2.097 |
T P |
| 1tq6_A |
402 |
444 |
57 |
2.71 |
1.75 |
8.306 |
2.026 |
T P |
| 1ega_B |
293 |
444 |
57 |
2.87 |
3.51 |
8.175 |
1.921 |
T P |
| 2zej_B |
162 |
444 |
49 |
2.80 |
4.08 |
7.223 |
1.691 |
T P |
| 2ve8_D |
68 |
444 |
42 |
2.42 |
4.76 |
6.743 |
1.665 |
T P |
| 2j5o_A |
72 |
444 |
40 |
2.34 |
5.00 |
6.718 |
1.638 |
T P |
| 2j5p_A |
70 |
444 |
42 |
2.36 |
2.38 |
6.705 |
1.708 |
T P |
| 1us8_A |
129 |
444 |
42 |
2.60 |
2.38 |
6.137 |
1.558 |
T P |
| 1ii8_A |
195 |
444 |
42 |
2.64 |
9.52 |
6.048 |
1.530 |
T P |
| 1f2u_A |
149 |
444 |
36 |
2.79 |
0.00 |
5.462 |
1.247 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]