LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_510.5wLII_11364_61.FR_1_700
Total number of 3D structures: 120
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
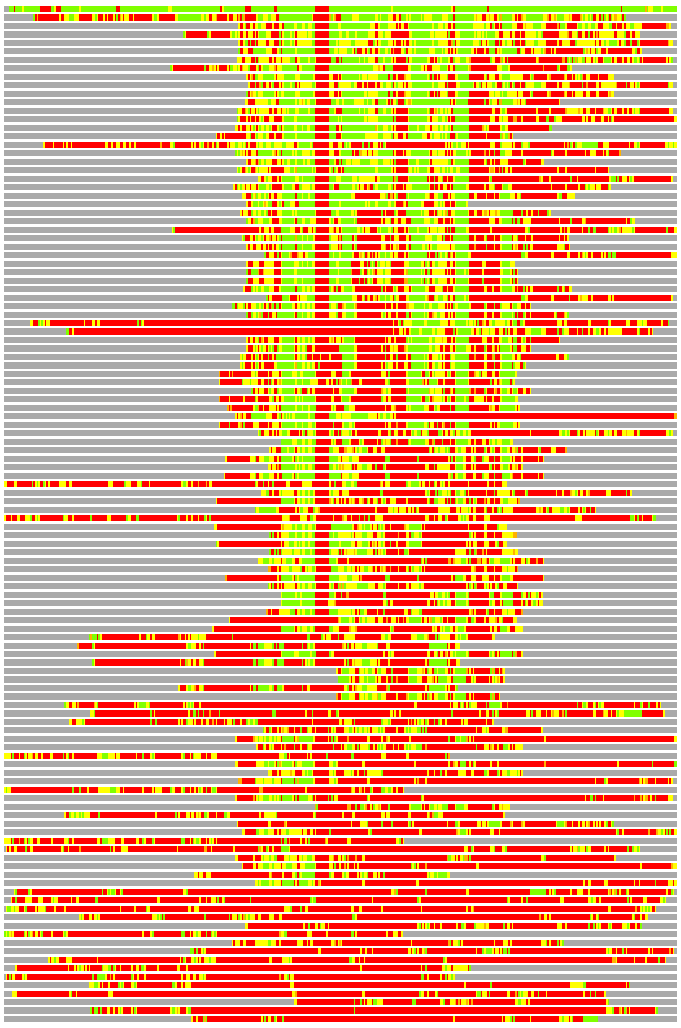
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 3e1s_A |
517 |
413 |
378 |
0.96 |
17.99 |
88.843 |
35.643 |
T P |
| 1w36_D |
538 |
413 |
249 |
2.31 |
18.07 |
44.688 |
10.348 |
T P |
| 2is6_A |
654 |
413 |
189 |
2.21 |
15.87 |
30.965 |
8.170 |
T P |
| 2gk6_A |
602 |
413 |
182 |
2.47 |
14.29 |
30.453 |
7.093 |
T P |
| 2pjr_F |
544 |
413 |
182 |
2.49 |
18.13 |
28.899 |
7.031 |
T P |
| 1pjr_A |
623 |
413 |
155 |
2.25 |
20.00 |
27.118 |
6.602 |
T P |
| 3g0h_A |
408 |
413 |
154 |
2.32 |
14.29 |
25.695 |
6.374 |
T P |
| 2eyq_A |
1146 |
413 |
138 |
2.18 |
13.77 |
25.662 |
6.044 |
T P |
| 1c4o_A |
504 |
413 |
147 |
2.37 |
17.01 |
25.211 |
5.963 |
T P |
| 1uaa_A |
636 |
413 |
156 |
2.55 |
16.67 |
25.028 |
5.893 |
T P |
| 1d2m_A |
552 |
413 |
148 |
2.33 |
17.57 |
24.973 |
6.078 |
T P |
| 3b85_A |
187 |
413 |
135 |
2.11 |
22.22 |
24.876 |
6.097 |
T P |
| 3fht_A |
392 |
413 |
147 |
2.30 |
12.93 |
24.477 |
6.113 |
T P |
| 2va8_B |
695 |
413 |
137 |
2.21 |
15.33 |
24.317 |
5.935 |
T P |
| 3ews_A |
416 |
413 |
123 |
2.09 |
17.07 |
23.453 |
5.610 |
T P |
| 2fwr_A |
434 |
413 |
117 |
2.16 |
12.82 |
22.771 |
5.180 |
T P |
| 1gm5_A |
729 |
413 |
151 |
2.68 |
10.60 |
22.557 |
5.425 |
T P |
| 1oyy_A |
512 |
413 |
135 |
2.41 |
9.63 |
21.845 |
5.376 |
T P |
| 1s2m_A |
377 |
413 |
121 |
2.27 |
13.22 |
21.182 |
5.105 |
T P |
| 2vso_A |
366 |
413 |
121 |
2.32 |
13.22 |
20.803 |
4.991 |
T P |
| 2v8o_A |
429 |
413 |
116 |
2.15 |
14.66 |
20.781 |
5.165 |
T P |
| 1oyw_A |
516 |
413 |
126 |
2.28 |
7.14 |
20.564 |
5.294 |
T P |
| 1l8q_A |
321 |
413 |
121 |
2.33 |
17.36 |
20.442 |
4.980 |
T P |
| 3b6e_A |
182 |
413 |
103 |
1.87 |
20.39 |
20.427 |
5.223 |
T P |
| 2hcb_D |
320 |
413 |
118 |
2.20 |
17.80 |
20.233 |
5.121 |
T P |
| 2qby_A |
366 |
413 |
120 |
2.33 |
15.00 |
19.117 |
4.945 |
T P |
| 1cu1_A |
645 |
413 |
121 |
2.40 |
13.22 |
19.081 |
4.838 |
T P |
| 1sxj_C |
322 |
413 |
109 |
2.29 |
12.84 |
18.409 |
4.555 |
T P |
| 1sxj_D |
328 |
413 |
113 |
2.44 |
11.50 |
18.133 |
4.445 |
T P |
| 1a1v_A |
432 |
413 |
112 |
2.39 |
16.96 |
18.004 |
4.496 |
T P |
| 2eyu_A |
247 |
413 |
106 |
2.23 |
12.26 |
17.978 |
4.553 |
T P |
| 2ewv_A |
343 |
413 |
108 |
2.27 |
12.04 |
17.905 |
4.566 |
T P |
| 2eww_A |
343 |
413 |
106 |
2.19 |
11.32 |
17.877 |
4.629 |
T P |
| 1sxj_A |
441 |
413 |
106 |
2.23 |
13.21 |
17.704 |
4.559 |
T P |
| 2f55_A |
432 |
413 |
105 |
2.26 |
16.19 |
17.610 |
4.447 |
T P |
| 1qvr_A |
803 |
413 |
113 |
2.41 |
14.16 |
17.567 |
4.504 |
T P |
| 1xxi_C |
366 |
413 |
106 |
2.23 |
12.26 |
17.479 |
4.550 |
T P |
| 1w36_B |
1158 |
413 |
105 |
2.54 |
13.33 |
16.973 |
3.974 |
T P |
| 1qhh_B |
261 |
413 |
99 |
2.60 |
17.17 |
16.784 |
3.671 |
T P |
| 1ojl_D |
297 |
413 |
105 |
2.40 |
13.33 |
16.625 |
4.201 |
T P |
| 1ny5_B |
385 |
413 |
103 |
2.38 |
11.65 |
16.621 |
4.156 |
T P |
| 1jr3_A |
366 |
413 |
98 |
2.10 |
11.22 |
16.618 |
4.455 |
T P |
| 1njg_A |
240 |
413 |
99 |
2.20 |
11.11 |
16.572 |
4.308 |
T P |
| 1s9h_A |
268 |
413 |
99 |
2.42 |
18.18 |
16.307 |
3.926 |
T P |
| 2hyd_A |
578 |
413 |
98 |
2.38 |
15.31 |
16.227 |
3.955 |
T P |
| 3dzd_A |
368 |
413 |
99 |
2.33 |
15.15 |
16.028 |
4.067 |
T P |
| 1u0j_A |
261 |
413 |
97 |
2.34 |
17.53 |
15.935 |
3.980 |
T P |
| 2gxa_E |
274 |
413 |
92 |
2.23 |
13.04 |
15.861 |
3.943 |
T P |
| 1qhh_A |
164 |
413 |
80 |
2.18 |
22.50 |
15.678 |
3.514 |
T P |
| 2v9p_D |
274 |
413 |
93 |
2.20 |
16.13 |
15.578 |
4.042 |
T P |
| 1hei_A |
443 |
413 |
108 |
2.73 |
7.41 |
15.406 |
3.812 |
T P |
| 1ye8_A |
172 |
413 |
86 |
2.19 |
25.58 |
14.768 |
3.750 |
T P |
| 2ffh_A |
407 |
413 |
90 |
2.58 |
13.33 |
14.664 |
3.356 |
T P |
| 2ncd_A |
358 |
413 |
84 |
2.21 |
8.33 |
14.378 |
3.632 |
T P |
| 3e70_C |
307 |
413 |
87 |
2.61 |
18.39 |
14.308 |
3.212 |
T P |
| 1n6m_A |
362 |
413 |
82 |
2.20 |
8.54 |
14.238 |
3.566 |
T P |
| 1fuu_B |
380 |
413 |
91 |
2.81 |
7.69 |
14.024 |
3.129 |
T P |
| 1ihu_A |
540 |
413 |
88 |
2.72 |
10.23 |
13.972 |
3.118 |
T P |
| 1g3q_A |
237 |
413 |
85 |
2.60 |
17.65 |
13.798 |
3.144 |
T P |
| 3bh1_D |
487 |
413 |
94 |
2.81 |
14.89 |
13.749 |
3.232 |
T P |
| 2qeq_A |
415 |
413 |
90 |
2.81 |
5.56 |
13.698 |
3.094 |
T P |
| 3dm5_A |
416 |
413 |
75 |
2.39 |
18.67 |
13.635 |
3.009 |
T P |
| 1jpn_A |
296 |
413 |
80 |
2.61 |
21.25 |
13.413 |
2.948 |
T P |
| 1rj9_B |
282 |
413 |
77 |
2.38 |
16.88 |
13.287 |
3.103 |
T P |
| 2j45_A |
297 |
413 |
81 |
2.75 |
18.52 |
13.195 |
2.845 |
T P |
| 1w9l_A |
726 |
413 |
79 |
2.46 |
12.66 |
13.126 |
3.085 |
T P |
| 3dm9_B |
305 |
413 |
79 |
2.82 |
18.99 |
13.055 |
2.706 |
T P |
| 1cz7_D |
365 |
413 |
70 |
2.16 |
10.00 |
12.800 |
3.093 |
T P |
| 2c03_B |
297 |
413 |
80 |
2.73 |
15.00 |
12.530 |
2.829 |
T P |
| 2zzy_A |
186 |
413 |
71 |
2.06 |
18.31 |
12.349 |
3.283 |
T P |
| 2zzz_A |
186 |
413 |
72 |
2.35 |
20.83 |
12.321 |
2.944 |
T P |
| 2px0_A |
258 |
413 |
70 |
2.61 |
18.57 |
12.272 |
2.580 |
T P |
| 1xzp_A |
456 |
413 |
75 |
2.62 |
13.33 |
12.043 |
2.757 |
T P |
| 1np6_B |
169 |
413 |
67 |
2.51 |
19.40 |
11.760 |
2.569 |
T P |
| 2ng1_A |
293 |
413 |
77 |
2.66 |
14.29 |
11.725 |
2.789 |
T P |
| 2j3e_A |
249 |
413 |
70 |
2.43 |
8.57 |
11.550 |
2.767 |
T P |
| 3a00_A |
153 |
413 |
67 |
2.15 |
20.90 |
11.518 |
2.982 |
T P |
| 3def_A |
244 |
413 |
69 |
2.45 |
5.80 |
11.452 |
2.708 |
T P |
| 821p_A |
166 |
413 |
68 |
2.49 |
19.12 |
11.298 |
2.626 |
T P |
| 2o52_A |
167 |
413 |
66 |
2.37 |
9.09 |
11.208 |
2.676 |
T P |
| 3bb3_A |
242 |
413 |
64 |
2.35 |
7.81 |
11.158 |
2.616 |
T P |
| 1jah_A |
166 |
413 |
66 |
2.40 |
18.18 |
11.144 |
2.636 |
T P |
| 1ump_A |
619 |
413 |
67 |
2.59 |
5.97 |
11.011 |
2.486 |
T P |
| 2owo_A |
586 |
413 |
68 |
2.51 |
10.29 |
10.796 |
2.610 |
T P |
| 2j28_9 |
430 |
413 |
68 |
2.87 |
10.29 |
10.740 |
2.286 |
T P |
| 1ls1_A |
289 |
413 |
66 |
2.81 |
7.58 |
10.594 |
2.266 |
T P |
| 1w9k_A |
719 |
413 |
61 |
2.47 |
11.48 |
10.421 |
2.378 |
T P |
| 2j7p_A |
292 |
413 |
67 |
2.69 |
10.45 |
10.386 |
2.403 |
T P |
| 1dgk_N |
898 |
413 |
69 |
2.73 |
8.70 |
10.313 |
2.435 |
T P |
| 1d1a_A |
745 |
413 |
61 |
2.51 |
4.92 |
10.254 |
2.337 |
T P |
| 2v3c_C |
403 |
413 |
70 |
2.77 |
7.14 |
10.210 |
2.442 |
T P |
| 1w9i_A |
702 |
413 |
58 |
2.57 |
17.24 |
10.201 |
2.175 |
T P |
| 1hkb_A |
899 |
413 |
62 |
2.79 |
8.06 |
10.081 |
2.146 |
T P |
| 1w9j_A |
728 |
413 |
58 |
2.43 |
15.52 |
9.977 |
2.290 |
T P |
| 2bgw_A |
219 |
413 |
54 |
2.61 |
3.70 |
9.896 |
1.993 |
T P |
| 1ffh_A |
287 |
413 |
63 |
2.76 |
9.52 |
9.792 |
2.205 |
T P |
| 2aka_A |
764 |
413 |
57 |
2.67 |
0.00 |
9.749 |
2.054 |
T P |
| 1fmv_A |
743 |
413 |
60 |
2.66 |
5.00 |
9.599 |
2.175 |
T P |
| 1bg3_A |
902 |
413 |
64 |
2.70 |
9.38 |
9.566 |
2.286 |
T P |
| 8ohm_A |
435 |
413 |
63 |
2.87 |
3.17 |
9.272 |
2.121 |
T P |
| 1lvk_A |
743 |
413 |
58 |
2.85 |
12.07 |
9.194 |
1.969 |
T P |
| 2jj9_A |
692 |
413 |
55 |
2.66 |
10.91 |
9.143 |
1.989 |
T P |
| 2bhn_A |
211 |
413 |
54 |
2.48 |
9.26 |
9.110 |
2.095 |
T P |
| 1jwy_A |
753 |
413 |
52 |
2.47 |
13.46 |
8.998 |
2.024 |
T P |
| 1qha_A |
903 |
413 |
53 |
2.58 |
5.66 |
8.991 |
1.974 |
T P |
| 1v9p_A |
584 |
413 |
56 |
2.69 |
7.14 |
8.906 |
2.004 |
T P |
| 1dgt_B |
660 |
413 |
56 |
2.79 |
7.14 |
8.833 |
1.941 |
T P |
| 1vom_A |
730 |
413 |
53 |
2.73 |
9.43 |
8.491 |
1.871 |
T P |
| 1ega_B |
293 |
413 |
54 |
2.79 |
7.41 |
8.340 |
1.869 |
T P |
| 1cza_N |
898 |
413 |
52 |
2.78 |
7.69 |
8.052 |
1.806 |
T P |
| 1p4d_B |
276 |
413 |
54 |
2.94 |
7.41 |
7.998 |
1.774 |
T P |
| 3ekj_A |
301 |
413 |
45 |
2.67 |
11.11 |
7.877 |
1.623 |
T P |
| 1yv3_A |
703 |
413 |
49 |
2.91 |
4.08 |
7.841 |
1.627 |
T P |
| 2dg8_B |
176 |
413 |
47 |
2.83 |
17.02 |
7.426 |
1.603 |
T P |
| 2a0i_A |
293 |
413 |
44 |
2.55 |
6.82 |
7.114 |
1.659 |
T P |
| 2cdm_A |
280 |
413 |
41 |
2.63 |
12.20 |
7.029 |
1.500 |
T P |
| 1omh_A |
282 |
413 |
44 |
2.88 |
11.36 |
6.864 |
1.478 |
T P |
| 2iyk_A |
155 |
413 |
40 |
2.73 |
7.50 |
6.841 |
1.415 |
T P |
| 1osb_A |
287 |
413 |
36 |
2.73 |
5.56 |
6.381 |
1.271 |
T P |
| 2q7t_A |
267 |
413 |
37 |
2.55 |
5.41 |
6.142 |
1.397 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]