LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_52.5wLII_10954_2
Total number of 3D structures: 79
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
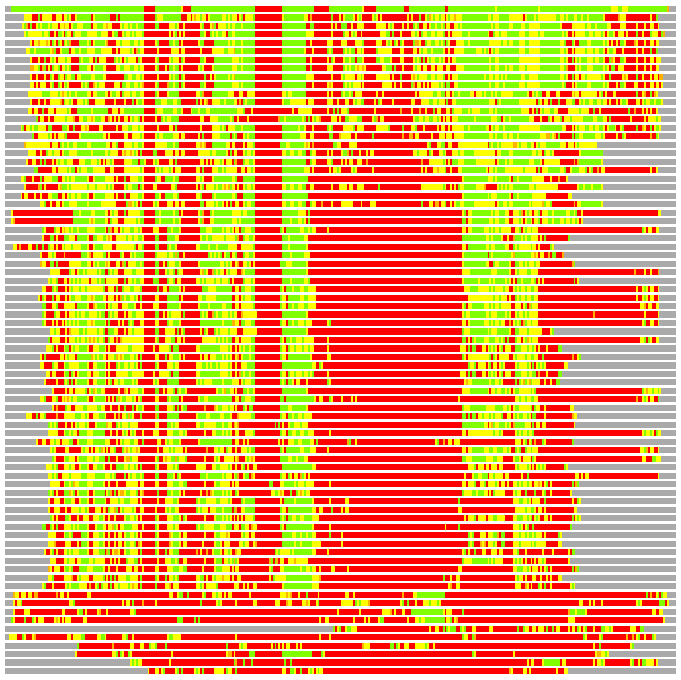
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1zyl_A |
325 |
343 |
294 |
0.95 |
18.03 |
83.395 |
27.942 |
T P |
| 2ppq_A |
310 |
343 |
219 |
2.21 |
12.79 |
44.222 |
9.468 |
T P |
| 2qg7_E |
369 |
343 |
216 |
2.27 |
11.11 |
41.381 |
9.095 |
T P |
| 3c5i_D |
355 |
343 |
219 |
2.35 |
7.76 |
41.284 |
8.949 |
T P |
| 3feg_A |
336 |
343 |
212 |
2.32 |
10.85 |
41.033 |
8.749 |
T P |
| 2ig7_B |
342 |
343 |
218 |
2.35 |
11.47 |
40.631 |
8.916 |
T P |
| 2cko_B |
346 |
343 |
209 |
2.27 |
11.00 |
40.295 |
8.803 |
T P |
| 3f2s_A |
354 |
343 |
210 |
2.35 |
10.95 |
40.268 |
8.569 |
T P |
| 3fi8_A |
339 |
343 |
209 |
2.30 |
8.61 |
39.970 |
8.700 |
T P |
| 2i7q_A |
344 |
343 |
206 |
2.38 |
11.17 |
38.941 |
8.294 |
T P |
| 3dxq_A |
294 |
343 |
197 |
2.39 |
14.72 |
36.669 |
7.917 |
T P |
| 3dxp_A |
329 |
343 |
201 |
2.44 |
9.45 |
36.655 |
7.907 |
T P |
| 2q83_A |
332 |
343 |
184 |
2.36 |
10.33 |
36.537 |
7.475 |
T P |
| 3d1u_A |
288 |
343 |
201 |
2.47 |
13.93 |
36.226 |
7.830 |
T P |
| 1nw1_A |
365 |
343 |
193 |
2.37 |
10.88 |
36.010 |
7.815 |
T P |
| 3csv_A |
322 |
343 |
182 |
2.30 |
8.79 |
35.442 |
7.574 |
T P |
| 2pyw_A |
417 |
343 |
187 |
2.52 |
13.37 |
34.098 |
7.134 |
T P |
| 2bkk_C |
262 |
343 |
185 |
2.39 |
10.27 |
33.589 |
7.433 |
T P |
| 1nd4_A |
255 |
343 |
171 |
2.45 |
8.19 |
32.997 |
6.694 |
T P |
| 2pul_A |
381 |
343 |
175 |
2.49 |
10.29 |
32.329 |
6.751 |
T P |
| 1tqp_A |
269 |
343 |
136 |
2.04 |
16.91 |
32.086 |
6.344 |
T P |
| 1l8t_A |
263 |
343 |
171 |
2.54 |
11.70 |
30.652 |
6.488 |
T P |
| 1zar_A |
267 |
343 |
132 |
2.09 |
15.15 |
30.628 |
6.025 |
T P |
| 1j7l_A |
263 |
343 |
171 |
2.52 |
11.70 |
30.390 |
6.522 |
T P |
| 3en9_A |
519 |
343 |
140 |
2.13 |
15.00 |
29.341 |
6.282 |
T P |
| 2vwb_A |
519 |
343 |
141 |
2.21 |
14.18 |
29.073 |
6.111 |
T P |
| 2jed_A |
326 |
343 |
138 |
2.39 |
13.04 |
25.562 |
5.543 |
T P |
| 3brb_B |
271 |
343 |
125 |
2.23 |
9.60 |
25.517 |
5.370 |
T P |
| 2pzi_A |
654 |
343 |
134 |
2.32 |
11.94 |
25.364 |
5.529 |
T P |
| 3e3p_A |
337 |
343 |
118 |
2.24 |
13.56 |
25.103 |
5.047 |
T P |
| 2j51_A |
288 |
343 |
124 |
2.33 |
13.71 |
25.056 |
5.112 |
T P |
| 2etr_A |
400 |
343 |
124 |
2.34 |
8.87 |
24.790 |
5.088 |
T P |
| 3g2f_A |
305 |
343 |
126 |
2.41 |
10.32 |
24.709 |
5.028 |
T P |
| 1phk_A |
277 |
343 |
132 |
2.41 |
5.30 |
24.529 |
5.256 |
T P |
| 1ql6_A |
281 |
343 |
132 |
2.40 |
5.30 |
24.521 |
5.278 |
T P |
| 1zrz_A |
312 |
343 |
131 |
2.41 |
12.21 |
24.454 |
5.227 |
T P |
| 2v55_C |
396 |
343 |
127 |
2.36 |
8.66 |
24.416 |
5.173 |
T P |
| 2z7r_A |
265 |
343 |
126 |
2.35 |
12.70 |
24.164 |
5.141 |
T P |
| 2f2u_A |
386 |
343 |
124 |
2.32 |
8.87 |
24.005 |
5.117 |
T P |
| 3daj_A |
245 |
343 |
130 |
2.58 |
5.38 |
23.966 |
4.855 |
T P |
| 2jfl_A |
288 |
343 |
126 |
2.43 |
7.94 |
23.540 |
4.974 |
T P |
| 2pml_X |
340 |
343 |
128 |
2.44 |
10.16 |
23.434 |
5.034 |
T P |
| 2w5a_A |
267 |
343 |
122 |
2.37 |
5.74 |
23.432 |
4.937 |
T P |
| 2ja0_A |
286 |
343 |
125 |
2.46 |
8.00 |
23.378 |
4.875 |
T P |
| 2h34_A |
243 |
343 |
119 |
2.29 |
9.24 |
23.368 |
4.982 |
T P |
| 2qo9_A |
277 |
343 |
115 |
2.44 |
12.17 |
23.254 |
4.523 |
T P |
| 2phk_A |
277 |
343 |
120 |
2.39 |
5.00 |
23.088 |
4.815 |
T P |
| 2qob_A |
281 |
343 |
109 |
2.41 |
10.09 |
22.750 |
4.347 |
T P |
| 3f69_B |
283 |
343 |
122 |
2.43 |
8.20 |
22.690 |
4.824 |
T P |
| 1k2p_A |
258 |
343 |
115 |
2.41 |
8.70 |
22.101 |
4.588 |
T P |
| 2p2i_A |
289 |
343 |
121 |
2.54 |
10.74 |
21.829 |
4.587 |
T P |
| 2buj_A |
291 |
343 |
111 |
2.45 |
6.31 |
21.601 |
4.361 |
T P |
| 2vrx_B |
275 |
343 |
116 |
2.62 |
11.21 |
21.554 |
4.269 |
T P |
| 2oh4_A |
299 |
343 |
113 |
2.53 |
7.08 |
21.291 |
4.292 |
T P |
| 2jav_A |
253 |
343 |
112 |
2.52 |
7.14 |
21.224 |
4.270 |
T P |
| 1fot_A |
300 |
343 |
111 |
2.44 |
9.91 |
21.044 |
4.365 |
T P |
| 1vr2_A |
275 |
343 |
118 |
2.57 |
6.78 |
21.018 |
4.417 |
T P |
| 3c7q_A |
291 |
343 |
107 |
2.50 |
9.35 |
20.582 |
4.122 |
T P |
| 1muo_A |
251 |
343 |
107 |
2.35 |
6.54 |
20.540 |
4.376 |
T P |
| 3d14_A |
262 |
343 |
109 |
2.48 |
3.67 |
20.515 |
4.230 |
T P |
| 2qoo_A |
273 |
343 |
112 |
2.58 |
9.82 |
20.290 |
4.181 |
T P |
| 3efw_B |
242 |
343 |
103 |
2.30 |
9.71 |
20.028 |
4.291 |
T P |
| 2qoq_A |
285 |
343 |
107 |
2.61 |
6.54 |
19.447 |
3.953 |
T P |
| 2qok_A |
281 |
343 |
108 |
2.66 |
7.41 |
19.392 |
3.906 |
T P |
| 1ywn_A |
281 |
343 |
102 |
2.63 |
5.88 |
19.265 |
3.741 |
T P |
| 3be2_A |
290 |
343 |
106 |
2.63 |
7.55 |
19.104 |
3.886 |
T P |
| 3cjf_A |
281 |
343 |
100 |
2.52 |
7.00 |
18.782 |
3.813 |
T P |
| 3cjg_A |
282 |
343 |
101 |
2.62 |
6.93 |
18.741 |
3.708 |
T P |
| 1y6b_A |
266 |
343 |
95 |
2.57 |
3.16 |
17.918 |
3.555 |
T P |
| 1qqc_A |
740 |
343 |
69 |
2.76 |
8.70 |
13.465 |
2.410 |
T P |
| 1r6b_X |
704 |
343 |
71 |
2.87 |
2.82 |
13.205 |
2.387 |
T P |
| 2dhr_A |
458 |
343 |
61 |
2.39 |
3.28 |
12.880 |
2.451 |
T P |
| 1g5a_A |
628 |
343 |
57 |
2.53 |
5.26 |
11.300 |
2.167 |
T P |
| 1ixz_A |
238 |
343 |
57 |
2.57 |
12.28 |
10.570 |
2.137 |
T P |
| 1jmx_B |
339 |
343 |
51 |
2.75 |
11.76 |
10.158 |
1.789 |
T P |
| 1iy2_A |
245 |
343 |
47 |
2.69 |
8.51 |
9.688 |
1.687 |
T P |
| 1via_A |
161 |
343 |
43 |
2.38 |
2.33 |
9.544 |
1.733 |
T P |
| 2iyv_A |
179 |
343 |
42 |
2.56 |
9.52 |
8.757 |
1.582 |
T P |
| 2g1k_A |
168 |
343 |
42 |
2.84 |
9.52 |
7.815 |
1.428 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]