LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_525.5wLII_11390_11
Total number of 3D structures: 47
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
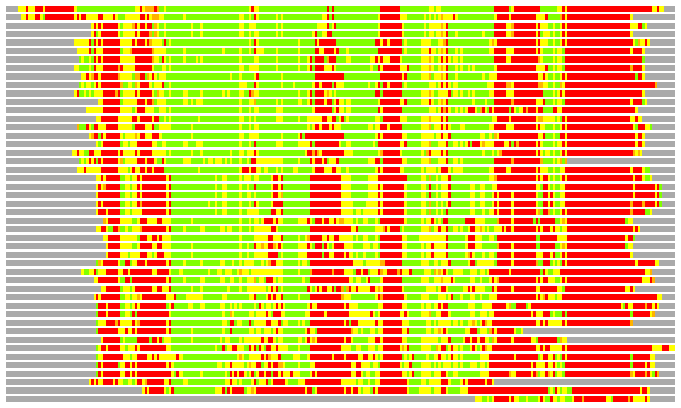
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1ypw_A |
692 |
275 |
183 |
1.69 |
15.85 |
63.509 |
10.235 |
T P |
| 1r7r_A |
683 |
275 |
173 |
2.01 |
15.61 |
54.022 |
8.189 |
T P |
| 3cf2_A |
659 |
275 |
160 |
1.96 |
13.12 |
50.237 |
7.778 |
T P |
| 1s3s_F |
441 |
275 |
155 |
1.97 |
13.55 |
48.641 |
7.472 |
T P |
| 3d8b_A |
281 |
275 |
156 |
2.04 |
13.46 |
47.752 |
7.304 |
T P |
| 3b9p_A |
268 |
275 |
151 |
1.95 |
13.91 |
46.802 |
7.371 |
T P |
| 3eie_A |
303 |
275 |
152 |
2.00 |
11.18 |
46.747 |
7.230 |
T P |
| 3eih_A |
319 |
275 |
156 |
2.02 |
11.54 |
46.365 |
7.347 |
T P |
| 2ce7_C |
421 |
275 |
146 |
2.04 |
14.38 |
46.118 |
6.822 |
T P |
| 1iy2_A |
245 |
275 |
154 |
2.08 |
12.34 |
45.045 |
7.054 |
T P |
| 1xwi_A |
322 |
275 |
155 |
2.07 |
13.55 |
44.563 |
7.154 |
T P |
| 2qp9_X |
288 |
275 |
147 |
2.05 |
12.24 |
43.345 |
6.837 |
T P |
| 1ixz_A |
238 |
275 |
148 |
2.10 |
12.84 |
43.152 |
6.731 |
T P |
| 1yqi_C |
708 |
275 |
152 |
2.13 |
11.18 |
42.441 |
6.826 |
T P |
| 2dhr_A |
458 |
275 |
162 |
2.11 |
10.49 |
42.433 |
7.318 |
T P |
| 2qz4_A |
223 |
275 |
138 |
2.06 |
11.59 |
42.259 |
6.396 |
T P |
| 2rko_A |
280 |
275 |
141 |
2.09 |
12.06 |
41.260 |
6.426 |
T P |
| 3cf0_A |
281 |
275 |
152 |
2.20 |
9.87 |
40.559 |
6.617 |
T P |
| 2zan_A |
312 |
275 |
148 |
2.14 |
15.54 |
39.086 |
6.600 |
T P |
| 1lv7_A |
251 |
275 |
150 |
2.16 |
11.33 |
38.869 |
6.637 |
T P |
| 1j7k_A |
299 |
275 |
131 |
2.11 |
12.98 |
37.614 |
5.922 |
T P |
| 1in5_A |
301 |
275 |
130 |
2.03 |
13.08 |
37.579 |
6.094 |
T P |
| 1in4_A |
298 |
275 |
130 |
2.07 |
13.85 |
37.516 |
5.987 |
T P |
| 1in8_A |
298 |
275 |
130 |
2.10 |
15.38 |
37.409 |
5.916 |
T P |
| 1in7_A |
298 |
275 |
134 |
2.16 |
14.93 |
35.422 |
5.922 |
T P |
| 1e94_F |
408 |
275 |
135 |
2.23 |
11.11 |
33.893 |
5.784 |
T P |
| 2c9o_A |
398 |
275 |
135 |
2.32 |
13.33 |
33.822 |
5.575 |
T P |
| 1g4a_E |
356 |
275 |
135 |
2.31 |
10.37 |
33.486 |
5.610 |
T P |
| 1g41_A |
334 |
275 |
134 |
2.23 |
11.19 |
33.427 |
5.762 |
T P |
| 1ofh_A |
309 |
275 |
131 |
2.22 |
11.45 |
33.332 |
5.647 |
T P |
| 1ixs_B |
315 |
275 |
128 |
2.26 |
10.94 |
33.264 |
5.430 |
T P |
| 2r62_A |
249 |
275 |
133 |
2.49 |
11.28 |
32.639 |
5.142 |
T P |
| 1ixr_C |
308 |
275 |
125 |
2.22 |
12.00 |
32.390 |
5.392 |
T P |
| 1doo_E |
393 |
275 |
130 |
2.42 |
10.00 |
32.102 |
5.151 |
T P |
| 1hqc_A |
314 |
275 |
129 |
2.27 |
11.63 |
31.654 |
5.448 |
T P |
| 1iqp_A |
326 |
275 |
128 |
2.24 |
14.06 |
31.509 |
5.481 |
T P |
| 2chq_A |
313 |
275 |
123 |
2.25 |
13.01 |
30.682 |
5.229 |
T P |
| 1njg_A |
240 |
275 |
122 |
2.41 |
13.11 |
30.212 |
4.859 |
T P |
| 2chg_A |
223 |
275 |
115 |
2.16 |
11.30 |
29.852 |
5.095 |
T P |
| 1um8_A |
327 |
275 |
122 |
2.27 |
12.30 |
29.769 |
5.139 |
T P |
| 1xxi_C |
366 |
275 |
124 |
2.39 |
12.90 |
29.439 |
4.971 |
T P |
| 1sxj_A |
441 |
275 |
124 |
2.50 |
12.10 |
28.910 |
4.764 |
T P |
| 1sxj_D |
328 |
275 |
122 |
2.41 |
17.21 |
28.842 |
4.853 |
T P |
| 1sxj_C |
322 |
275 |
114 |
2.51 |
14.04 |
26.732 |
4.363 |
T P |
| 2qgz_A |
183 |
275 |
103 |
2.27 |
15.53 |
25.193 |
4.341 |
T P |
| 1rkb_A |
173 |
275 |
78 |
2.32 |
17.95 |
19.079 |
3.219 |
T P |
| 2dvw_B |
73 |
275 |
43 |
2.58 |
13.95 |
10.186 |
1.604 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]