LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_532.5wLII_11390_53
Total number of 3D structures: 71
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
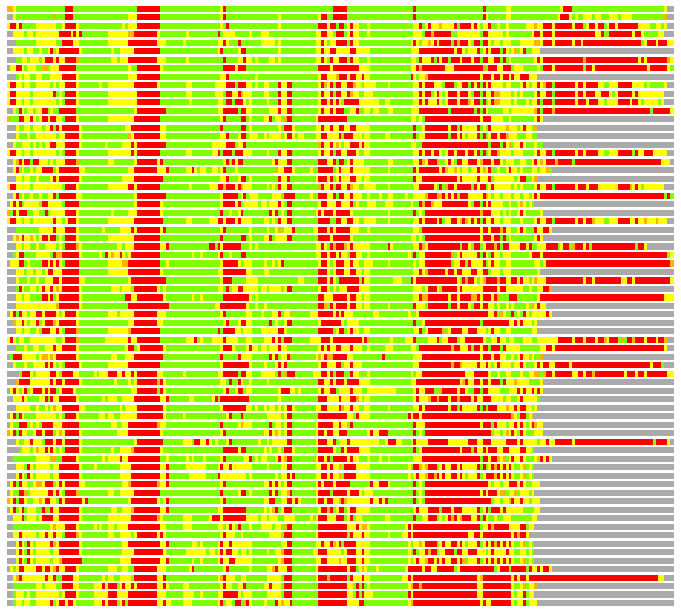
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 3f4k_A |
254 |
231 |
207 |
0.84 |
19.32 |
88.614 |
21.964 |
T P |
| 3e7p_A |
253 |
231 |
203 |
1.37 |
17.24 |
82.980 |
13.844 |
T P |
| 3bus_A |
251 |
231 |
171 |
2.11 |
14.04 |
58.214 |
7.740 |
T P |
| 2o57_A |
282 |
231 |
165 |
2.16 |
12.73 |
54.147 |
7.305 |
T P |
| 1xxl_A |
234 |
231 |
155 |
2.18 |
14.19 |
52.394 |
6.787 |
T P |
| 1ve3_B |
226 |
231 |
137 |
1.94 |
12.41 |
51.743 |
6.723 |
T P |
| 1vl5_A |
231 |
231 |
143 |
2.04 |
13.29 |
50.751 |
6.674 |
T P |
| 1p91_A |
268 |
231 |
130 |
1.82 |
20.00 |
50.597 |
6.757 |
T P |
| 3dlc_A |
219 |
231 |
129 |
1.89 |
13.95 |
49.998 |
6.495 |
T P |
| 2fk8_A |
281 |
231 |
165 |
2.30 |
10.30 |
49.462 |
6.883 |
T P |
| 1tpy_A |
285 |
231 |
164 |
2.32 |
12.80 |
49.355 |
6.791 |
T P |
| 1kpi_A |
291 |
231 |
161 |
2.30 |
9.32 |
49.235 |
6.718 |
T P |
| 2p35_A |
245 |
231 |
131 |
1.94 |
18.32 |
49.098 |
6.406 |
T P |
| 2nxc_A |
249 |
231 |
127 |
1.90 |
20.47 |
49.052 |
6.345 |
T P |
| 3d2l_C |
242 |
231 |
136 |
2.05 |
16.18 |
48.184 |
6.313 |
T P |
| 1y8c_A |
246 |
231 |
138 |
2.05 |
17.39 |
48.150 |
6.417 |
T P |
| 2pxx_A |
213 |
231 |
128 |
1.96 |
17.19 |
48.127 |
6.199 |
T P |
| 1l1e_A |
272 |
231 |
156 |
2.30 |
12.18 |
48.037 |
6.488 |
T P |
| 2gh1_A |
281 |
231 |
143 |
2.18 |
14.69 |
48.014 |
6.269 |
T P |
| 1qzz_A |
340 |
231 |
142 |
2.23 |
13.38 |
47.850 |
6.105 |
T P |
| 1wzn_A |
244 |
231 |
133 |
1.94 |
15.04 |
47.836 |
6.513 |
T P |
| 1kph_B |
285 |
231 |
160 |
2.36 |
11.88 |
47.745 |
6.517 |
T P |
| 2p7i_B |
229 |
231 |
139 |
2.06 |
16.55 |
47.700 |
6.437 |
T P |
| 3dtn_A |
220 |
231 |
136 |
2.04 |
14.71 |
47.607 |
6.364 |
T P |
| 2b3t_A |
276 |
231 |
130 |
1.85 |
20.00 |
47.483 |
6.653 |
T P |
| 1kpg_A |
285 |
231 |
164 |
2.41 |
12.20 |
47.480 |
6.529 |
T P |
| 1o54_A |
265 |
231 |
130 |
1.97 |
14.62 |
47.452 |
6.265 |
T P |
| 3cjt_A |
254 |
231 |
124 |
1.94 |
20.97 |
47.444 |
6.091 |
T P |
| 3ege_A |
247 |
231 |
137 |
2.09 |
12.41 |
47.253 |
6.259 |
T P |
| 2p8j_B |
207 |
231 |
138 |
2.04 |
11.59 |
46.832 |
6.443 |
T P |
| 3g5l_A |
222 |
231 |
139 |
2.10 |
13.67 |
46.440 |
6.311 |
T P |
| 2avn_A |
247 |
231 |
132 |
2.06 |
17.42 |
46.035 |
6.119 |
T P |
| 2yqz_A |
261 |
231 |
128 |
2.00 |
13.28 |
45.655 |
6.086 |
T P |
| 1ri5_A |
252 |
231 |
140 |
2.22 |
12.86 |
45.517 |
6.025 |
T P |
| 2gs9_A |
211 |
231 |
124 |
1.93 |
15.32 |
45.210 |
6.119 |
T P |
| 1vlm_A |
207 |
231 |
127 |
2.04 |
20.47 |
45.141 |
5.927 |
T P |
| 2as0_A |
396 |
231 |
131 |
2.09 |
20.61 |
44.928 |
5.977 |
T P |
| 2pjd_A |
334 |
231 |
119 |
1.99 |
15.13 |
44.912 |
5.706 |
T P |
| 3bkw_A |
219 |
231 |
134 |
2.08 |
18.66 |
44.841 |
6.137 |
T P |
| 1nkv_C |
249 |
231 |
156 |
2.49 |
16.03 |
44.773 |
6.019 |
T P |
| 2ex4_A |
221 |
231 |
138 |
2.10 |
15.94 |
44.656 |
6.267 |
T P |
| 1t43_A |
274 |
231 |
127 |
2.09 |
19.69 |
44.574 |
5.787 |
T P |
| 3e23_A |
198 |
231 |
137 |
2.10 |
13.14 |
44.457 |
6.240 |
T P |
| 3ccf_B |
242 |
231 |
134 |
2.28 |
15.67 |
44.332 |
5.622 |
T P |
| 3dh0_B |
190 |
231 |
127 |
2.15 |
15.75 |
44.315 |
5.651 |
T P |
| 1im8_A |
225 |
231 |
136 |
2.21 |
12.50 |
43.416 |
5.888 |
T P |
| 3dli_A |
221 |
231 |
130 |
2.21 |
13.08 |
43.392 |
5.622 |
T P |
| 3bxo_A |
236 |
231 |
132 |
2.25 |
12.88 |
43.350 |
5.612 |
T P |
| 2yxe_A |
214 |
231 |
116 |
2.08 |
19.83 |
42.483 |
5.317 |
T P |
| 1dus_A |
194 |
231 |
124 |
1.96 |
12.90 |
41.683 |
6.029 |
T P |
| 1sg9_A |
274 |
231 |
122 |
2.03 |
13.93 |
41.572 |
5.733 |
T P |
| 3bkx_A |
274 |
231 |
136 |
2.40 |
11.03 |
41.435 |
5.450 |
T P |
| 3cgg_A |
186 |
231 |
117 |
2.05 |
16.24 |
41.311 |
5.454 |
T P |
| 1l3i_A |
186 |
231 |
128 |
2.09 |
19.53 |
41.298 |
5.832 |
T P |
| 1d2g_A |
292 |
231 |
135 |
2.35 |
17.78 |
41.212 |
5.521 |
T P |
| 1r8x_B |
284 |
231 |
135 |
2.38 |
17.78 |
41.200 |
5.438 |
T P |
| 1nv8_A |
271 |
231 |
118 |
2.03 |
14.41 |
41.176 |
5.552 |
T P |
| 3dmg_A |
371 |
231 |
122 |
2.02 |
10.66 |
41.134 |
5.758 |
T P |
| 1vq1_A |
266 |
231 |
122 |
2.14 |
16.39 |
40.915 |
5.447 |
T P |
| 3e8s_A |
220 |
231 |
127 |
2.14 |
15.75 |
40.604 |
5.677 |
T P |
| 3cc8_A |
211 |
231 |
132 |
2.18 |
12.12 |
40.231 |
5.792 |
T P |
| 1dl5_A |
317 |
231 |
119 |
2.16 |
18.49 |
40.155 |
5.267 |
T P |
| 1i1n_A |
224 |
231 |
113 |
2.10 |
18.58 |
39.996 |
5.133 |
T P |
| 1xva_A |
292 |
231 |
133 |
2.35 |
18.05 |
39.918 |
5.433 |
T P |
| 1r74_B |
279 |
231 |
134 |
2.34 |
15.67 |
39.729 |
5.484 |
T P |
| 2azt_B |
277 |
231 |
134 |
2.38 |
15.67 |
39.553 |
5.397 |
T P |
| 1g6q_1 |
328 |
231 |
115 |
2.10 |
16.52 |
38.801 |
5.231 |
T P |
| 1r18_A |
223 |
231 |
114 |
2.15 |
13.16 |
37.337 |
5.062 |
T P |
| 1jg1_A |
215 |
231 |
115 |
2.14 |
13.91 |
35.659 |
5.136 |
T P |
| 1vbf_B |
226 |
231 |
110 |
2.23 |
13.64 |
33.681 |
4.721 |
T P |
| 2pbf_A |
218 |
231 |
111 |
2.23 |
15.32 |
32.615 |
4.760 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]