LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_57.5wLII_10954_40
Total number of 3D structures: 72
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
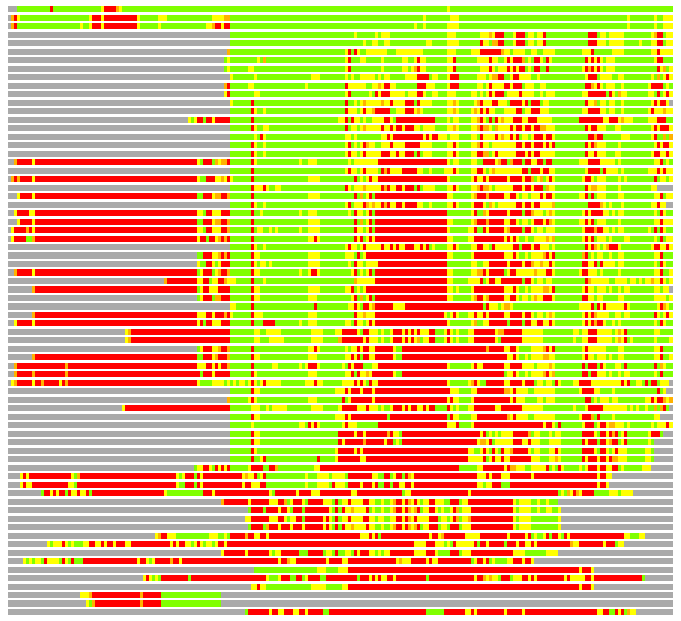
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 3cio_A |
255 |
221 |
213 |
0.59 |
23.94 |
95.884 |
30.724 |
T P |
| 2ved_B |
254 |
221 |
205 |
1.42 |
20.49 |
87.757 |
13.463 |
T P |
| 3bfv_A |
241 |
221 |
201 |
1.36 |
21.39 |
86.100 |
13.807 |
T P |
| 1ion_A |
243 |
221 |
135 |
1.71 |
16.30 |
56.833 |
7.454 |
T P |
| 1g3q_A |
237 |
221 |
135 |
1.69 |
18.52 |
56.775 |
7.542 |
T P |
| 1hyq_A |
232 |
221 |
137 |
1.86 |
18.98 |
55.442 |
6.990 |
T P |
| 1wcv_1 |
243 |
221 |
133 |
1.73 |
19.55 |
54.781 |
7.283 |
T P |
| 2bek_A |
244 |
221 |
134 |
1.78 |
18.66 |
51.721 |
7.124 |
T P |
| 3ea0_B |
242 |
221 |
132 |
1.96 |
16.67 |
44.965 |
6.413 |
T P |
| 2oze_A |
284 |
221 |
126 |
1.94 |
23.02 |
44.196 |
6.188 |
T P |
| 3fkq_A |
354 |
221 |
127 |
1.96 |
14.96 |
44.114 |
6.179 |
T P |
| 3end_A |
268 |
221 |
124 |
2.04 |
16.13 |
42.837 |
5.806 |
T P |
| 1g20_H |
262 |
221 |
124 |
2.14 |
16.13 |
40.215 |
5.542 |
T P |
| 1ihu_A |
540 |
221 |
111 |
2.00 |
18.92 |
39.377 |
5.290 |
T P |
| 1de0_A |
289 |
221 |
123 |
2.27 |
17.07 |
39.150 |
5.192 |
T P |
| 2ph1_A |
247 |
221 |
118 |
2.10 |
19.49 |
39.146 |
5.368 |
T P |
| 1cp2_A |
269 |
221 |
122 |
2.17 |
14.75 |
39.060 |
5.375 |
T P |
| 1xdb_A |
289 |
221 |
121 |
2.19 |
16.53 |
39.012 |
5.294 |
T P |
| 3dm5_A |
416 |
221 |
112 |
2.09 |
18.75 |
38.850 |
5.122 |
T P |
| 2c8v_A |
271 |
221 |
120 |
2.16 |
16.67 |
38.806 |
5.309 |
T P |
| 3dm9_B |
305 |
221 |
113 |
2.10 |
18.58 |
38.769 |
5.132 |
T P |
| 2afh_E |
289 |
221 |
120 |
2.29 |
16.67 |
38.524 |
5.025 |
T P |
| 1rj9_B |
282 |
221 |
112 |
1.99 |
22.32 |
38.237 |
5.362 |
T P |
| 1rw4_A |
271 |
221 |
119 |
2.15 |
16.81 |
38.155 |
5.288 |
T P |
| 1qzx_A |
425 |
221 |
110 |
1.98 |
19.09 |
38.149 |
5.281 |
T P |
| 2j7p_A |
292 |
221 |
111 |
1.99 |
22.52 |
38.015 |
5.319 |
T P |
| 1j8m_F |
295 |
221 |
110 |
2.07 |
19.09 |
37.916 |
5.070 |
T P |
| 2j37_W |
479 |
221 |
112 |
2.06 |
19.64 |
37.808 |
5.197 |
T P |
| 1xd8_A |
289 |
221 |
117 |
2.23 |
15.38 |
37.723 |
5.031 |
T P |
| 1jpn_A |
296 |
221 |
109 |
2.00 |
22.94 |
37.544 |
5.191 |
T P |
| 2j28_9 |
430 |
221 |
110 |
2.06 |
13.64 |
37.302 |
5.094 |
T P |
| 1ls1_A |
289 |
221 |
113 |
2.18 |
21.24 |
36.903 |
4.955 |
T P |
| 1zu4_A |
305 |
221 |
116 |
2.33 |
18.10 |
36.899 |
4.766 |
T P |
| 2ng1_A |
293 |
221 |
110 |
2.10 |
23.64 |
36.889 |
5.005 |
T P |
| 2j45_A |
297 |
221 |
111 |
2.07 |
22.52 |
36.493 |
5.114 |
T P |
| 1yrb_B |
260 |
221 |
104 |
2.01 |
11.54 |
35.947 |
4.930 |
T P |
| 1fts_A |
295 |
221 |
110 |
2.23 |
20.91 |
35.827 |
4.722 |
T P |
| 1j8y_F |
291 |
221 |
103 |
2.00 |
18.45 |
35.799 |
4.916 |
T P |
| 1eg7_A |
549 |
221 |
116 |
2.27 |
16.38 |
35.631 |
4.894 |
T P |
| 1fpm_A |
549 |
221 |
115 |
2.25 |
15.65 |
35.236 |
4.900 |
T P |
| 2ffh_A |
407 |
221 |
103 |
2.04 |
21.36 |
35.221 |
4.810 |
T P |
| 2c03_B |
297 |
221 |
104 |
2.11 |
22.12 |
35.198 |
4.707 |
T P |
| 2v3c_C |
403 |
221 |
105 |
2.22 |
18.10 |
34.697 |
4.524 |
T P |
| 1ffh_A |
287 |
221 |
101 |
2.12 |
23.76 |
34.564 |
4.551 |
T P |
| 3e70_C |
307 |
221 |
110 |
2.41 |
14.55 |
33.740 |
4.382 |
T P |
| 2qm7_A |
314 |
221 |
97 |
1.93 |
17.53 |
33.720 |
4.786 |
T P |
| 2p67_A |
302 |
221 |
96 |
2.03 |
11.46 |
33.423 |
4.507 |
T P |
| 3do6_A |
543 |
221 |
111 |
2.34 |
14.41 |
33.257 |
4.548 |
T P |
| 2qm8_A |
320 |
221 |
94 |
1.94 |
18.09 |
33.071 |
4.617 |
T P |
| 3cwq_A |
206 |
221 |
88 |
1.92 |
18.18 |
31.472 |
4.365 |
T P |
| 2ax4_A |
198 |
221 |
84 |
2.03 |
10.71 |
28.984 |
3.939 |
T P |
| 2yvu_A |
179 |
221 |
86 |
2.03 |
13.95 |
28.933 |
4.042 |
T P |
| 2gks_B |
529 |
221 |
83 |
2.00 |
15.66 |
28.482 |
3.960 |
T P |
| 3cr8_A |
493 |
221 |
80 |
2.05 |
17.50 |
27.493 |
3.717 |
T P |
| 1qvr_A |
803 |
221 |
78 |
2.22 |
19.23 |
25.406 |
3.360 |
T P |
| 3cwg_B |
507 |
221 |
63 |
2.54 |
3.17 |
19.585 |
2.383 |
T P |
| 1bg1_A |
559 |
221 |
59 |
2.30 |
5.08 |
19.491 |
2.461 |
T P |
| 1f5n_A |
570 |
221 |
55 |
2.44 |
5.45 |
18.342 |
2.169 |
T P |
| 2v1d_A |
666 |
221 |
60 |
2.63 |
3.33 |
17.953 |
2.194 |
T P |
| 2iw5_A |
666 |
221 |
59 |
2.64 |
3.39 |
17.889 |
2.151 |
T P |
| 2h94_A |
647 |
221 |
62 |
2.70 |
4.84 |
17.749 |
2.212 |
T P |
| 2z3y_A |
643 |
221 |
57 |
2.58 |
3.51 |
17.383 |
2.124 |
T P |
| 1cii_A |
602 |
221 |
58 |
2.57 |
10.34 |
17.375 |
2.169 |
T P |
| 2hko_A |
647 |
221 |
62 |
2.90 |
1.61 |
17.261 |
2.065 |
T P |
| 2dw4_A |
634 |
221 |
54 |
2.49 |
5.56 |
16.799 |
2.089 |
T P |
| 1zvu_A |
685 |
221 |
51 |
2.69 |
3.92 |
14.853 |
1.825 |
T P |
| 1m1j_D |
194 |
221 |
33 |
1.55 |
3.03 |
14.058 |
1.996 |
T P |
| 2i1j_A |
481 |
221 |
45 |
2.62 |
2.22 |
13.364 |
1.653 |
T P |
| 1ei3_A |
159 |
221 |
33 |
1.99 |
3.03 |
13.003 |
1.581 |
T P |
| 2v71_A |
160 |
221 |
25 |
1.74 |
12.00 |
10.684 |
1.358 |
T P |
| 1c1g_A |
284 |
221 |
24 |
1.90 |
12.50 |
10.288 |
1.203 |
T P |
| 1n3k_A |
130 |
221 |
29 |
2.45 |
6.90 |
9.133 |
1.136 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]