LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_58.5wLII_10954_58
Total number of 3D structures: 73
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
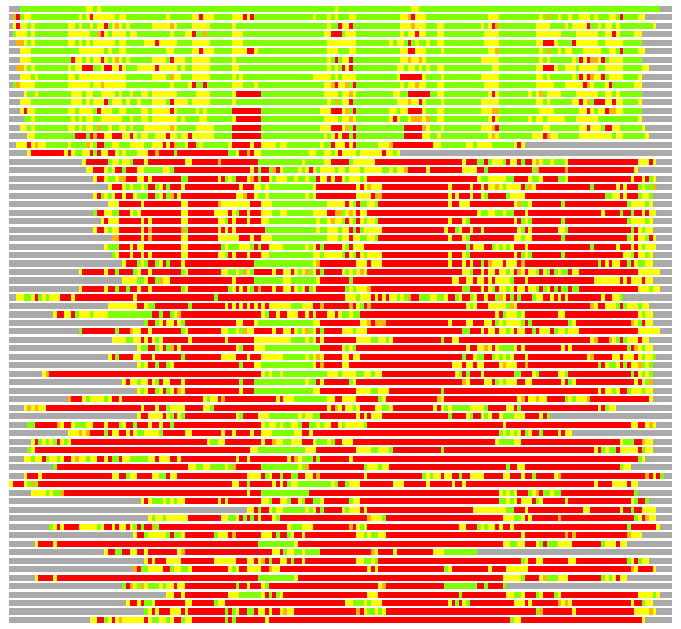
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1yk9_A |
184 |
181 |
175 |
0.63 |
15.43 |
95.660 |
23.861 |
T P |
| 1cul_B |
190 |
181 |
166 |
1.93 |
14.46 |
77.185 |
8.164 |
T P |
| 2w01_A |
201 |
181 |
170 |
1.99 |
15.88 |
76.699 |
8.124 |
T P |
| 3et6_B |
183 |
181 |
165 |
2.01 |
15.15 |
75.672 |
7.808 |
T P |
| 1wc1_A |
196 |
181 |
165 |
2.12 |
17.58 |
74.694 |
7.423 |
T P |
| 1azs_B |
189 |
181 |
165 |
1.96 |
13.94 |
72.430 |
8.023 |
T P |
| 1cs4_A |
189 |
181 |
162 |
2.04 |
14.20 |
72.372 |
7.561 |
T P |
| 1wc3_A |
198 |
181 |
161 |
2.02 |
17.39 |
71.638 |
7.599 |
T P |
| 1azs_A |
190 |
181 |
161 |
2.10 |
14.29 |
71.521 |
7.322 |
T P |
| 3et6_A |
184 |
181 |
166 |
2.09 |
15.66 |
71.049 |
7.571 |
T P |
| 1fx2_A |
235 |
181 |
155 |
1.93 |
13.55 |
70.603 |
7.631 |
T P |
| 1cul_A |
189 |
181 |
161 |
2.06 |
15.53 |
70.441 |
7.458 |
T P |
| 1ybt_B |
172 |
181 |
149 |
2.01 |
17.45 |
69.104 |
7.053 |
T P |
| 1fx4_A |
231 |
181 |
160 |
2.08 |
15.00 |
66.768 |
7.329 |
T P |
| 1ybu_A |
166 |
181 |
150 |
1.99 |
17.33 |
64.587 |
7.190 |
T P |
| 1y10_C |
363 |
181 |
140 |
2.06 |
15.00 |
58.292 |
6.485 |
T P |
| 1w25_A |
454 |
181 |
110 |
2.14 |
13.64 |
42.466 |
4.902 |
T P |
| 3b60_A |
572 |
181 |
63 |
2.33 |
7.94 |
25.538 |
2.597 |
T P |
| 2fb8_A |
259 |
181 |
64 |
2.44 |
4.69 |
24.346 |
2.524 |
T P |
| 2fr7_A |
397 |
181 |
64 |
2.59 |
12.50 |
23.295 |
2.378 |
T P |
| 2jkq_A |
258 |
181 |
63 |
2.50 |
4.76 |
23.063 |
2.423 |
T P |
| 3c4c_A |
258 |
181 |
59 |
2.53 |
15.25 |
22.930 |
2.240 |
T P |
| 2j0k_B |
612 |
181 |
60 |
2.60 |
5.00 |
22.814 |
2.224 |
T P |
| 2ofu_A |
273 |
181 |
62 |
2.63 |
8.06 |
22.466 |
2.274 |
T P |
| 2pl0_A |
268 |
181 |
60 |
2.46 |
10.00 |
22.278 |
2.342 |
T P |
| 3bym_A |
272 |
181 |
58 |
2.44 |
6.90 |
21.730 |
2.282 |
T P |
| 1o6y_A |
260 |
181 |
58 |
2.44 |
6.90 |
21.595 |
2.281 |
T P |
| 3lck_A |
271 |
181 |
59 |
2.55 |
5.08 |
21.539 |
2.229 |
T P |
| 3bys_A |
255 |
181 |
57 |
2.50 |
10.53 |
21.427 |
2.193 |
T P |
| 1qpc_A |
271 |
181 |
58 |
2.45 |
10.34 |
21.423 |
2.272 |
T P |
| 2ijm_B |
261 |
181 |
54 |
2.53 |
9.26 |
21.316 |
2.055 |
T P |
| 1uwj_A |
264 |
181 |
63 |
2.73 |
4.76 |
21.239 |
2.226 |
T P |
| 1mru_B |
271 |
181 |
57 |
2.44 |
3.51 |
21.122 |
2.248 |
T P |
| 1uwh_A |
264 |
181 |
56 |
2.58 |
3.57 |
21.040 |
2.087 |
T P |
| 2qp2_A |
498 |
181 |
57 |
2.69 |
5.26 |
21.024 |
2.040 |
T P |
| 2zm1_A |
271 |
181 |
59 |
2.86 |
6.78 |
20.950 |
1.991 |
T P |
| 1ys7_B |
226 |
181 |
57 |
2.51 |
8.77 |
20.730 |
2.186 |
T P |
| 3bz3_A |
259 |
181 |
54 |
2.58 |
7.41 |
20.701 |
2.013 |
T P |
| 3d4q_A |
264 |
181 |
62 |
2.79 |
4.84 |
20.468 |
2.144 |
T P |
| 2ofv_A |
244 |
181 |
56 |
2.65 |
7.14 |
20.398 |
2.037 |
T P |
| 1qpd_A |
271 |
181 |
52 |
2.39 |
13.46 |
20.106 |
2.092 |
T P |
| 3f61_A |
271 |
181 |
55 |
2.70 |
3.64 |
20.026 |
1.967 |
T P |
| 2j0l_A |
276 |
181 |
52 |
2.50 |
7.69 |
19.968 |
2.002 |
T P |
| 3f69_B |
283 |
181 |
51 |
2.30 |
3.92 |
19.846 |
2.129 |
T P |
| 2of2_A |
271 |
181 |
51 |
2.40 |
11.76 |
19.801 |
2.037 |
T P |
| 1mp8_A |
252 |
181 |
50 |
2.33 |
8.00 |
19.706 |
2.057 |
T P |
| 2v8o_A |
429 |
181 |
53 |
2.72 |
1.89 |
19.569 |
1.876 |
T P |
| 2ayx_A |
254 |
181 |
49 |
2.66 |
6.12 |
19.220 |
1.777 |
T P |
| 2jko_A |
256 |
181 |
48 |
2.46 |
12.50 |
18.652 |
1.874 |
T P |
| 1ihu_A |
540 |
181 |
48 |
2.66 |
6.25 |
18.441 |
1.741 |
T P |
| 1b0u_A |
258 |
181 |
47 |
2.68 |
4.26 |
17.991 |
1.692 |
T P |
| 1dyq_A |
228 |
181 |
46 |
2.63 |
13.04 |
17.438 |
1.683 |
T P |
| 2j0j_A |
611 |
181 |
44 |
2.37 |
4.55 |
17.172 |
1.780 |
T P |
| 1pf4_A |
520 |
181 |
45 |
2.63 |
4.44 |
17.100 |
1.647 |
T P |
| 2jvk_A |
132 |
181 |
40 |
2.29 |
5.00 |
17.096 |
1.672 |
T P |
| 2hyd_A |
578 |
181 |
47 |
2.88 |
6.38 |
16.413 |
1.577 |
T P |
| 1w36_B |
1158 |
181 |
41 |
2.43 |
4.88 |
16.275 |
1.623 |
T P |
| 1mvo_A |
121 |
181 |
37 |
2.14 |
8.11 |
16.124 |
1.652 |
T P |
| 2ayz_A |
133 |
181 |
41 |
2.45 |
4.88 |
15.869 |
1.610 |
T P |
| 2jkm_A |
260 |
181 |
43 |
2.64 |
6.98 |
15.858 |
1.569 |
T P |
| 2jvj_A |
132 |
181 |
39 |
2.60 |
7.69 |
14.769 |
1.445 |
T P |
| 1g29_1 |
372 |
181 |
40 |
2.74 |
10.00 |
14.518 |
1.409 |
T P |
| 1srr_C |
121 |
181 |
38 |
2.66 |
5.26 |
14.513 |
1.375 |
T P |
| 2a9o_A |
117 |
181 |
34 |
2.35 |
5.88 |
14.499 |
1.388 |
T P |
| 2jvi_A |
132 |
181 |
35 |
2.45 |
14.29 |
14.222 |
1.370 |
T P |
| 2iyn_B |
124 |
181 |
38 |
2.83 |
5.26 |
14.032 |
1.297 |
T P |
| 1nat_A |
119 |
181 |
37 |
2.85 |
5.41 |
13.895 |
1.254 |
T P |
| 2a9r_A |
117 |
181 |
33 |
2.39 |
6.06 |
13.559 |
1.324 |
T P |
| 2p08_B |
110 |
181 |
32 |
2.26 |
6.25 |
13.542 |
1.353 |
T P |
| 1f51_E |
119 |
181 |
35 |
2.67 |
5.71 |
13.343 |
1.264 |
T P |
| 2og8_B |
249 |
181 |
36 |
2.80 |
0.00 |
13.168 |
1.240 |
T P |
| 1mb3_A |
117 |
181 |
35 |
2.92 |
2.86 |
13.145 |
1.160 |
T P |
| 2p04_A |
107 |
181 |
29 |
2.59 |
6.90 |
11.349 |
1.076 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]