LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_59.5wLII_10954_62
Total number of 3D structures: 39
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
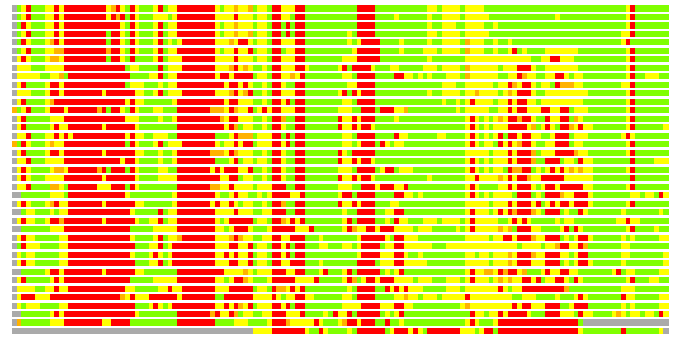
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 2iko_A |
338 |
139 |
107 |
1.96 |
11.21 |
65.459 |
5.192 |
T P |
| 2i4q_A |
336 |
139 |
106 |
1.92 |
13.21 |
65.417 |
5.247 |
T P |
| 2g24_A |
329 |
139 |
104 |
1.86 |
11.54 |
61.818 |
5.307 |
T P |
| 1hrn_A |
334 |
139 |
105 |
1.91 |
10.48 |
61.042 |
5.227 |
T P |
| 3d91_B |
337 |
139 |
105 |
1.99 |
13.33 |
60.438 |
5.016 |
T P |
| 1smr_A |
331 |
139 |
103 |
2.16 |
10.68 |
58.565 |
4.548 |
T P |
| 1lya_B |
241 |
139 |
104 |
2.22 |
13.46 |
56.613 |
4.492 |
T P |
| 3cms_A |
320 |
139 |
103 |
2.28 |
13.59 |
51.937 |
4.336 |
T P |
| 1uh9_A |
325 |
139 |
97 |
2.25 |
17.53 |
51.687 |
4.126 |
T P |
| 1tzs_A |
322 |
139 |
96 |
2.17 |
6.25 |
50.485 |
4.236 |
T P |
| 1flh_A |
326 |
139 |
99 |
2.26 |
6.06 |
50.260 |
4.196 |
T P |
| 3psg_A |
365 |
139 |
95 |
2.10 |
13.68 |
50.025 |
4.322 |
T P |
| 1dpj_A |
329 |
139 |
97 |
2.26 |
7.22 |
49.517 |
4.114 |
T P |
| 4cms_A |
320 |
139 |
95 |
2.11 |
14.74 |
49.386 |
4.291 |
T P |
| 1psa_A |
326 |
139 |
94 |
2.13 |
13.83 |
48.839 |
4.215 |
T P |
| 1yg9_A |
330 |
139 |
94 |
2.04 |
9.57 |
48.809 |
4.398 |
T P |
| 1htr_B |
329 |
139 |
95 |
2.08 |
8.42 |
48.647 |
4.357 |
T P |
| 5pep_A |
326 |
139 |
94 |
2.26 |
12.77 |
48.402 |
3.988 |
T P |
| 4pep_A |
326 |
139 |
96 |
2.12 |
12.50 |
48.222 |
4.316 |
T P |
| 1fq5_A |
329 |
139 |
97 |
2.33 |
7.22 |
47.907 |
4.000 |
T P |
| 1qrp_E |
326 |
139 |
94 |
2.26 |
10.64 |
47.765 |
3.978 |
T P |
| 1g0v_A |
329 |
139 |
94 |
2.20 |
8.51 |
47.724 |
4.083 |
T P |
| 2apr_A |
325 |
139 |
92 |
2.06 |
14.13 |
47.638 |
4.253 |
T P |
| 1f34_A |
326 |
139 |
93 |
2.18 |
12.90 |
47.593 |
4.088 |
T P |
| 2qzx_A |
342 |
139 |
93 |
2.07 |
7.53 |
47.571 |
4.292 |
T P |
| 1am5_A |
324 |
139 |
93 |
2.21 |
9.68 |
47.420 |
4.032 |
T P |
| 1od1_A |
330 |
139 |
93 |
2.24 |
11.83 |
46.919 |
3.980 |
T P |
| 2h6t_A |
340 |
139 |
93 |
2.33 |
5.38 |
46.453 |
3.828 |
T P |
| 1j71_A |
334 |
139 |
96 |
2.41 |
7.29 |
45.914 |
3.821 |
T P |
| 1zap_A |
341 |
139 |
93 |
2.29 |
5.38 |
45.547 |
3.898 |
T P |
| 1eag_A |
339 |
139 |
91 |
2.29 |
6.59 |
45.441 |
3.805 |
T P |
| 1jgf_A |
328 |
139 |
90 |
2.27 |
11.11 |
45.267 |
3.805 |
T P |
| 1oew_A |
328 |
139 |
92 |
2.36 |
10.87 |
45.224 |
3.734 |
T P |
| 2ewy_A |
374 |
139 |
92 |
2.34 |
13.04 |
45.130 |
3.773 |
T P |
| 2asi_A |
356 |
139 |
90 |
2.40 |
4.44 |
44.969 |
3.600 |
T P |
| 2qzw_A |
341 |
139 |
91 |
2.33 |
6.59 |
44.354 |
3.750 |
T P |
| 2anl_A |
327 |
139 |
89 |
2.20 |
8.99 |
44.170 |
3.873 |
T P |
| 1b5f_A |
239 |
139 |
72 |
2.10 |
15.28 |
39.845 |
3.275 |
T P |
| 2id5_B |
473 |
139 |
42 |
2.11 |
4.76 |
22.742 |
1.903 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]