LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_64.5wLII_10961_20
Total number of 3D structures: 58
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
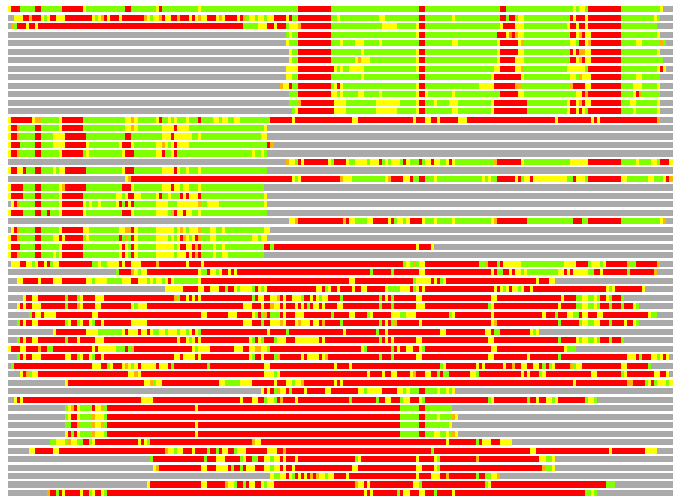
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1ys7_B |
226 |
220 |
176 |
0.84 |
15.34 |
78.911 |
18.633 |
T P |
| 2gwr_A |
225 |
220 |
135 |
2.08 |
18.52 |
50.089 |
6.194 |
T P |
| 2oqr_A |
226 |
220 |
105 |
1.80 |
18.10 |
43.979 |
5.533 |
T P |
| 2d1v_A |
103 |
220 |
94 |
1.51 |
14.89 |
41.246 |
5.852 |
T P |
| 1gxq_A |
105 |
220 |
95 |
1.64 |
18.95 |
40.780 |
5.460 |
T P |
| 2z9m_A |
100 |
220 |
92 |
1.55 |
13.04 |
40.038 |
5.567 |
T P |
| 2hwv_A |
101 |
220 |
93 |
1.66 |
15.05 |
39.779 |
5.275 |
T P |
| 2pmu_B |
103 |
220 |
95 |
1.78 |
13.68 |
39.707 |
5.054 |
T P |
| 1kgs_A |
219 |
220 |
92 |
1.51 |
14.13 |
39.569 |
5.714 |
T P |
| 1p2f_A |
217 |
220 |
90 |
1.72 |
17.78 |
38.505 |
4.958 |
T P |
| 1qqi_A |
104 |
220 |
91 |
1.64 |
19.78 |
38.341 |
5.240 |
T P |
| 1odd_A |
100 |
220 |
86 |
1.88 |
18.60 |
35.327 |
4.354 |
T P |
| 1opc_A |
99 |
220 |
83 |
1.72 |
19.28 |
34.946 |
4.557 |
T P |
| 1w25_A |
454 |
220 |
85 |
2.13 |
20.00 |
32.377 |
3.812 |
T P |
| 1zes_A |
121 |
220 |
74 |
1.77 |
20.27 |
31.039 |
3.956 |
T P |
| 2eub_A |
121 |
220 |
72 |
1.79 |
15.28 |
30.098 |
3.801 |
T P |
| 1zh2_A |
120 |
220 |
71 |
1.78 |
19.72 |
29.944 |
3.769 |
T P |
| 1xhe_B |
122 |
220 |
70 |
1.66 |
18.57 |
29.735 |
3.975 |
T P |
| 2hqn_A |
109 |
220 |
88 |
2.12 |
14.77 |
29.521 |
3.962 |
T P |
| 1xhf_B |
122 |
220 |
70 |
1.71 |
17.14 |
29.287 |
3.871 |
T P |
| 2hqr_A |
223 |
220 |
87 |
2.15 |
12.64 |
29.236 |
3.861 |
T P |
| 2a9o_A |
117 |
220 |
68 |
1.65 |
19.12 |
29.021 |
3.881 |
T P |
| 1mvo_A |
121 |
220 |
67 |
1.59 |
17.91 |
28.923 |
3.964 |
T P |
| 1b00_A |
122 |
220 |
72 |
1.97 |
18.06 |
28.850 |
3.472 |
T P |
| 2a9r_A |
117 |
220 |
67 |
1.61 |
19.40 |
28.742 |
3.920 |
T P |
| 2jpb_A |
104 |
220 |
75 |
1.99 |
18.67 |
28.427 |
3.595 |
T P |
| 2iyn_B |
124 |
220 |
75 |
2.07 |
17.33 |
26.410 |
3.453 |
T P |
| 2jb9_B |
122 |
220 |
71 |
1.95 |
15.49 |
25.388 |
3.457 |
T P |
| 2jba_A |
125 |
220 |
70 |
1.87 |
15.71 |
25.355 |
3.561 |
T P |
| 2jba_B |
121 |
220 |
69 |
1.99 |
15.94 |
24.635 |
3.306 |
T P |
| 1h6k_C |
733 |
220 |
72 |
2.55 |
8.33 |
22.017 |
2.719 |
T P |
| 1lrz_A |
400 |
220 |
50 |
2.49 |
2.00 |
16.412 |
1.931 |
T P |
| 1bg1_A |
559 |
220 |
55 |
2.64 |
1.82 |
15.911 |
2.011 |
T P |
| 2h94_A |
647 |
220 |
54 |
2.73 |
1.85 |
15.820 |
1.905 |
T P |
| 2hko_A |
647 |
220 |
50 |
2.61 |
8.00 |
15.617 |
1.845 |
T P |
| 2z3y_A |
643 |
220 |
51 |
2.71 |
9.80 |
15.232 |
1.816 |
T P |
| 1ha0_A |
494 |
220 |
54 |
2.74 |
1.85 |
15.215 |
1.904 |
T P |
| 2dw4_A |
634 |
220 |
53 |
2.81 |
7.55 |
15.020 |
1.824 |
T P |
| 1y1u_A |
544 |
220 |
49 |
2.66 |
4.08 |
14.896 |
1.779 |
T P |
| 2v1d_A |
666 |
220 |
49 |
2.70 |
10.20 |
14.853 |
1.749 |
T P |
| 3cwg_B |
507 |
220 |
48 |
2.76 |
6.25 |
14.606 |
1.677 |
T P |
| 2iw5_A |
666 |
220 |
48 |
2.90 |
10.42 |
14.042 |
1.599 |
T P |
| 1f5n_A |
570 |
220 |
45 |
2.79 |
15.56 |
13.069 |
1.557 |
T P |
| 1qvr_A |
803 |
220 |
39 |
2.92 |
2.56 |
12.134 |
1.290 |
T P |
| 1jch_A |
468 |
220 |
38 |
2.88 |
5.26 |
11.821 |
1.274 |
T P |
| 2ve7_B |
303 |
220 |
35 |
2.44 |
2.86 |
11.376 |
1.380 |
T P |
| 2jz6_A |
77 |
220 |
36 |
2.59 |
2.78 |
10.808 |
1.341 |
T P |
| 2b9c_B |
142 |
220 |
28 |
2.00 |
7.14 |
10.622 |
1.331 |
T P |
| 2fxo_A |
129 |
220 |
29 |
2.28 |
0.00 |
10.536 |
1.220 |
T P |
| 2fxm_A |
126 |
220 |
28 |
2.09 |
0.00 |
10.439 |
1.281 |
T P |
| 1c1g_A |
284 |
220 |
30 |
2.37 |
3.33 |
10.182 |
1.214 |
T P |
| 2j01_1 |
88 |
220 |
28 |
2.44 |
3.57 |
9.212 |
1.102 |
T P |
| 2b5u_A |
470 |
220 |
31 |
2.97 |
3.23 |
9.129 |
1.011 |
T P |
| 2qam_Z |
77 |
220 |
27 |
2.55 |
0.00 |
8.807 |
1.019 |
T P |
| 2i2t_X |
77 |
220 |
30 |
2.64 |
3.33 |
8.802 |
1.095 |
T P |
| 2jl6_1 |
87 |
220 |
28 |
2.85 |
10.71 |
8.526 |
0.950 |
T P |
| 3bbo_Y |
76 |
220 |
26 |
2.64 |
0.00 |
7.490 |
0.948 |
T P |
| 3f1f_1 |
88 |
220 |
21 |
2.65 |
9.52 |
7.252 |
0.763 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]