LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_66.5wLII_10961_25
Total number of 3D structures: 76
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
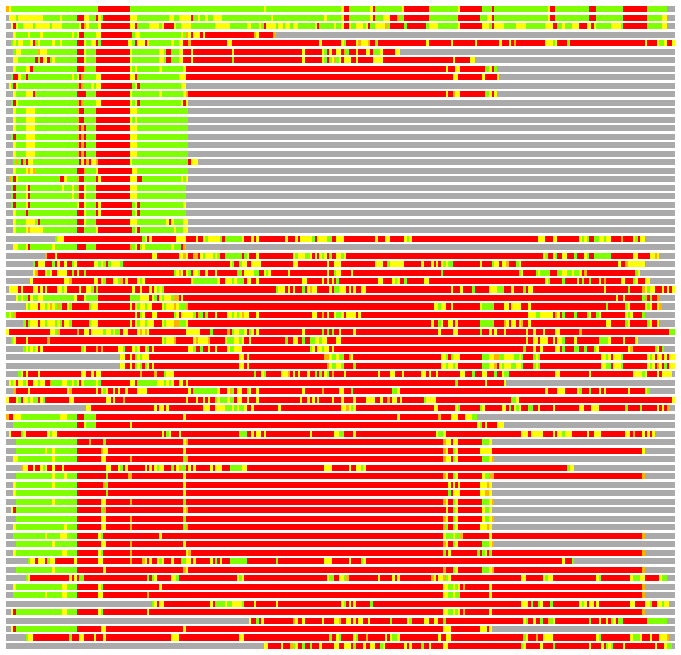
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1hfe_L |
396 |
275 |
221 |
0.78 |
23.08 |
79.604 |
24.972 |
T P |
| 3c8y_A |
574 |
275 |
214 |
1.65 |
18.22 |
71.699 |
12.251 |
T P |
| 1e08_A |
371 |
275 |
206 |
2.18 |
22.82 |
51.377 |
9.032 |
T P |
| 1jnr_B |
149 |
275 |
65 |
1.92 |
18.46 |
21.264 |
3.213 |
T P |
| 2fug_3 |
737 |
275 |
84 |
2.45 |
11.90 |
21.255 |
3.292 |
T P |
| 2v4j_B |
380 |
275 |
70 |
2.15 |
17.14 |
20.442 |
3.111 |
T P |
| 3c7b_B |
363 |
275 |
69 |
2.12 |
14.49 |
20.319 |
3.103 |
T P |
| 2fgo_A |
81 |
275 |
60 |
1.65 |
20.00 |
20.305 |
3.426 |
T P |
| 1jb0_C |
80 |
275 |
59 |
1.75 |
27.12 |
20.046 |
3.196 |
T P |
| 2vkr_A |
103 |
275 |
57 |
1.68 |
21.05 |
19.807 |
3.208 |
T P |
| 1blu_A |
80 |
275 |
61 |
1.81 |
18.03 |
19.789 |
3.197 |
T P |
| 1xer_A |
102 |
275 |
57 |
1.68 |
21.05 |
19.694 |
3.198 |
T P |
| 1a6l_A |
106 |
275 |
56 |
1.64 |
17.86 |
19.424 |
3.221 |
T P |
| 1ftc_A |
106 |
275 |
56 |
1.66 |
17.86 |
19.351 |
3.180 |
T P |
| 1b0v_A |
106 |
275 |
56 |
1.76 |
19.64 |
19.136 |
3.007 |
T P |
| 2fd2_A |
106 |
275 |
56 |
1.73 |
17.86 |
19.131 |
3.062 |
T P |
| 1g6b_A |
106 |
275 |
56 |
1.77 |
17.86 |
19.115 |
3.001 |
T P |
| 1frk_A |
106 |
275 |
56 |
1.76 |
17.86 |
19.086 |
3.008 |
T P |
| 2fug_9 |
154 |
275 |
57 |
1.80 |
21.05 |
19.034 |
3.005 |
T P |
| 1h98_A |
77 |
275 |
55 |
1.69 |
16.36 |
18.944 |
3.078 |
T P |
| 2z8q_A |
66 |
275 |
54 |
1.77 |
22.22 |
18.549 |
2.890 |
T P |
| 1fca_A |
55 |
275 |
54 |
1.57 |
24.07 |
18.405 |
3.229 |
T P |
| 2fdn_A |
55 |
275 |
54 |
1.56 |
25.93 |
18.340 |
3.263 |
T P |
| 1dur_A |
55 |
275 |
53 |
1.37 |
20.75 |
18.282 |
3.594 |
T P |
| 1fdx |
54 |
275 |
52 |
1.27 |
23.08 |
17.935 |
3.794 |
T P |
| 1bc6_A |
77 |
275 |
53 |
1.84 |
18.87 |
17.888 |
2.727 |
T P |
| 1bqx_A |
77 |
275 |
54 |
1.87 |
20.37 |
17.784 |
2.741 |
T P |
| 2iv2_X |
696 |
275 |
74 |
2.59 |
6.76 |
17.441 |
2.750 |
T P |
| 1clf_A |
55 |
275 |
50 |
1.70 |
24.00 |
16.745 |
2.785 |
T P |
| 2vpz_A |
734 |
275 |
67 |
2.56 |
5.97 |
16.479 |
2.516 |
T P |
| 1yqt_A |
515 |
275 |
71 |
2.83 |
5.63 |
16.136 |
2.421 |
T P |
| 2v3v_A |
721 |
275 |
62 |
2.57 |
8.06 |
16.133 |
2.321 |
T P |
| 1h0h_A |
976 |
275 |
67 |
2.53 |
7.46 |
16.063 |
2.547 |
T P |
| 3cf4_A |
766 |
275 |
69 |
2.94 |
10.14 |
15.876 |
2.269 |
T P |
| 1k0t_A |
80 |
275 |
55 |
2.28 |
16.36 |
15.516 |
2.314 |
T P |
| 1h7x_B |
1019 |
275 |
66 |
2.73 |
4.55 |
15.504 |
2.328 |
T P |
| 1jfl_A |
228 |
275 |
66 |
2.74 |
3.03 |
15.228 |
2.324 |
T P |
| 1gte_D |
1014 |
275 |
67 |
2.82 |
4.48 |
14.828 |
2.291 |
T P |
| 2nap_A |
720 |
275 |
65 |
2.72 |
4.62 |
14.811 |
2.301 |
T P |
| 2dx7_A |
228 |
275 |
57 |
2.73 |
1.75 |
14.333 |
2.014 |
T P |
| 1xdw_A |
331 |
275 |
56 |
2.57 |
3.57 |
14.297 |
2.095 |
T P |
| 2h88_B |
239 |
275 |
65 |
2.74 |
7.69 |
14.113 |
2.287 |
T P |
| 1zoy_B |
239 |
275 |
62 |
2.62 |
6.45 |
14.111 |
2.282 |
T P |
| 3bk7_A |
593 |
275 |
62 |
2.94 |
4.84 |
14.099 |
2.042 |
T P |
| 2o01_C |
80 |
275 |
55 |
2.32 |
18.18 |
13.808 |
2.269 |
T P |
| 1kqf_A |
981 |
275 |
55 |
2.79 |
7.27 |
13.415 |
1.904 |
T P |
| 2nx9_B |
453 |
275 |
58 |
2.76 |
12.07 |
13.335 |
2.025 |
T P |
| 1q16_B |
509 |
275 |
56 |
2.72 |
1.79 |
13.287 |
1.983 |
T P |
| 1kqf_B |
289 |
275 |
43 |
2.17 |
18.60 |
13.173 |
1.896 |
T P |
| 2vpz_B |
193 |
275 |
38 |
1.61 |
15.79 |
12.529 |
2.217 |
T P |
| 1y4z_B |
509 |
275 |
53 |
2.82 |
7.55 |
12.472 |
1.812 |
T P |
| 1fdd_A |
106 |
275 |
38 |
2.09 |
15.79 |
11.870 |
1.736 |
T P |
| 1frh_A |
106 |
275 |
39 |
2.19 |
20.51 |
11.864 |
1.703 |
T P |
| 1pc4_A |
106 |
275 |
41 |
2.34 |
12.20 |
11.805 |
1.682 |
T P |
| 2c3m_A |
1231 |
275 |
49 |
2.73 |
4.08 |
11.773 |
1.729 |
T P |
| 1f5c_A |
106 |
275 |
39 |
2.37 |
17.95 |
11.766 |
1.580 |
T P |
| 1f5b_A |
106 |
275 |
37 |
2.01 |
18.92 |
11.686 |
1.751 |
T P |
| 1pc5_A |
106 |
275 |
37 |
1.96 |
13.51 |
11.657 |
1.799 |
T P |
| 1d3w_A |
106 |
275 |
38 |
2.19 |
18.42 |
11.634 |
1.657 |
T P |
| 1b0t_A |
106 |
275 |
37 |
1.84 |
16.22 |
11.601 |
1.911 |
T P |
| 1frl_A |
106 |
275 |
38 |
2.21 |
18.42 |
11.599 |
1.644 |
T P |
| 1frj_A |
106 |
275 |
40 |
2.26 |
15.00 |
11.598 |
1.695 |
T P |
| 1fri_A |
106 |
275 |
39 |
1.92 |
15.38 |
11.553 |
1.931 |
T P |
| 1g3o_A |
106 |
275 |
38 |
2.18 |
18.42 |
11.543 |
1.665 |
T P |
| 1gao_A |
106 |
275 |
38 |
2.19 |
15.79 |
11.538 |
1.661 |
T P |
| 2c42_A |
1231 |
275 |
44 |
2.58 |
2.27 |
11.495 |
1.641 |
T P |
| 7fd1_A |
106 |
275 |
37 |
2.32 |
18.92 |
11.484 |
1.526 |
T P |
| 2bs2_B |
239 |
275 |
49 |
2.56 |
10.20 |
11.419 |
1.841 |
T P |
| 1fd2_A |
106 |
275 |
39 |
1.91 |
15.38 |
11.409 |
1.940 |
T P |
| 1frx_A |
106 |
275 |
38 |
1.81 |
15.79 |
11.328 |
1.993 |
T P |
| 2ivf_B |
337 |
275 |
47 |
2.67 |
0.00 |
11.250 |
1.696 |
T P |
| 1ff2_A |
106 |
275 |
39 |
2.03 |
12.82 |
11.231 |
1.829 |
T P |
| 2bs3_B |
239 |
275 |
46 |
2.66 |
6.52 |
11.225 |
1.670 |
T P |
| 1frm_A |
106 |
275 |
36 |
1.89 |
16.67 |
11.181 |
1.807 |
T P |
| 1kf6_B |
243 |
275 |
46 |
2.88 |
4.35 |
11.053 |
1.543 |
T P |
| 1nek_B |
238 |
275 |
40 |
2.73 |
12.50 |
10.011 |
1.415 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]