LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_68.5wLII_10961_32
Total number of 3D structures: 71
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
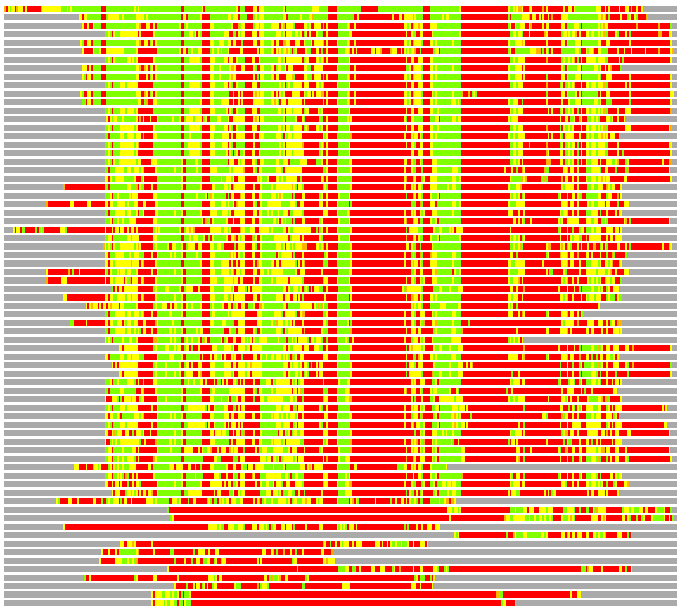
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1qvr_A |
803 |
388 |
260 |
1.64 |
20.00 |
63.264 |
14.947 |
T P |
| 1r6b_X |
704 |
388 |
221 |
1.97 |
17.65 |
49.480 |
10.664 |
T P |
| 1ofh_A |
309 |
388 |
180 |
2.32 |
13.89 |
32.005 |
7.428 |
T P |
| 1ixs_B |
315 |
388 |
169 |
2.20 |
10.65 |
31.988 |
7.347 |
T P |
| 1doo_E |
393 |
388 |
177 |
2.19 |
15.82 |
31.836 |
7.745 |
T P |
| 1um8_A |
327 |
388 |
180 |
2.27 |
13.89 |
31.650 |
7.592 |
T P |
| 1j7k_A |
299 |
388 |
163 |
2.13 |
11.04 |
31.289 |
7.324 |
T P |
| 1im2_A |
346 |
388 |
173 |
2.15 |
13.29 |
31.232 |
7.679 |
T P |
| 1e94_F |
408 |
388 |
171 |
2.14 |
16.37 |
31.216 |
7.639 |
T P |
| 1in7_A |
298 |
388 |
161 |
2.19 |
11.18 |
30.964 |
7.025 |
T P |
| 1g4a_E |
356 |
388 |
169 |
2.13 |
14.20 |
30.824 |
7.579 |
T P |
| 1g41_A |
334 |
388 |
169 |
2.10 |
14.20 |
30.648 |
7.692 |
T P |
| 1in5_A |
301 |
388 |
161 |
2.14 |
11.18 |
29.339 |
7.172 |
T P |
| 2c9o_A |
398 |
388 |
153 |
2.22 |
11.76 |
29.326 |
6.582 |
T P |
| 1in6_A |
300 |
388 |
161 |
2.18 |
10.56 |
29.263 |
7.068 |
T P |
| 3d8b_A |
281 |
388 |
163 |
2.16 |
14.11 |
29.229 |
7.200 |
T P |
| 1in4_A |
298 |
388 |
158 |
2.19 |
11.39 |
29.077 |
6.907 |
T P |
| 1in8_A |
298 |
388 |
156 |
2.11 |
11.54 |
28.751 |
7.048 |
T P |
| 1ixr_C |
308 |
388 |
170 |
2.23 |
11.18 |
28.716 |
7.293 |
T P |
| 3eie_A |
303 |
388 |
156 |
2.27 |
10.26 |
27.682 |
6.592 |
T P |
| 2qz4_A |
223 |
388 |
153 |
2.24 |
7.84 |
27.449 |
6.543 |
T P |
| 2dhr_A |
458 |
388 |
158 |
2.25 |
11.39 |
27.409 |
6.710 |
T P |
| 2chg_A |
223 |
388 |
150 |
2.09 |
12.00 |
27.152 |
6.836 |
T P |
| 1s3s_F |
441 |
388 |
160 |
2.27 |
11.88 |
27.091 |
6.753 |
T P |
| 1iy2_A |
245 |
388 |
150 |
2.25 |
11.33 |
26.878 |
6.395 |
T P |
| 1iqp_A |
326 |
388 |
147 |
2.12 |
11.56 |
26.629 |
6.636 |
T P |
| 3dzd_A |
368 |
388 |
157 |
2.46 |
15.92 |
26.354 |
6.141 |
T P |
| 2chq_A |
313 |
388 |
154 |
2.30 |
12.34 |
26.224 |
6.426 |
T P |
| 1hqc_A |
314 |
388 |
155 |
2.49 |
9.03 |
26.178 |
5.976 |
T P |
| 3b9p_A |
268 |
388 |
143 |
2.19 |
11.19 |
26.049 |
6.241 |
T P |
| 3cf0_A |
281 |
388 |
158 |
2.39 |
12.03 |
25.810 |
6.346 |
T P |
| 1yqi_C |
708 |
388 |
157 |
2.37 |
11.46 |
25.775 |
6.344 |
T P |
| 3cf2_A |
659 |
388 |
154 |
2.40 |
12.99 |
25.584 |
6.169 |
T P |
| 2c9c_A |
244 |
388 |
154 |
2.42 |
15.58 |
25.552 |
6.115 |
T P |
| 1r7r_A |
683 |
388 |
153 |
2.36 |
11.11 |
25.521 |
6.223 |
T P |
| 2r44_A |
330 |
388 |
143 |
2.22 |
15.38 |
25.358 |
6.159 |
T P |
| 3eih_A |
319 |
388 |
145 |
2.20 |
11.72 |
25.069 |
6.293 |
T P |
| 1ypw_A |
692 |
388 |
145 |
2.29 |
12.41 |
24.987 |
6.061 |
T P |
| 1xwi_A |
322 |
388 |
146 |
2.27 |
9.59 |
24.868 |
6.157 |
T P |
| 1g8p_A |
321 |
388 |
142 |
2.15 |
13.38 |
24.822 |
6.319 |
T P |
| 2bjw_A |
242 |
388 |
149 |
2.42 |
16.78 |
24.787 |
5.902 |
T P |
| 2zan_A |
312 |
388 |
146 |
2.41 |
10.27 |
24.745 |
5.818 |
T P |
| 2vii_A |
247 |
388 |
153 |
2.45 |
18.30 |
24.696 |
6.010 |
T P |
| 2bjv_A |
236 |
388 |
144 |
2.34 |
18.75 |
24.579 |
5.903 |
T P |
| 1sxj_D |
328 |
388 |
140 |
2.22 |
14.29 |
24.525 |
6.047 |
T P |
| 1ixz_A |
238 |
388 |
134 |
2.21 |
11.94 |
24.317 |
5.812 |
T P |
| 2ce7_C |
421 |
388 |
142 |
2.33 |
14.08 |
24.298 |
5.850 |
T P |
| 1jr3_A |
366 |
388 |
146 |
2.35 |
10.27 |
24.297 |
5.957 |
T P |
| 2qp9_X |
288 |
388 |
145 |
2.27 |
11.03 |
24.270 |
6.110 |
T P |
| 1xxi_C |
366 |
388 |
146 |
2.39 |
10.96 |
24.179 |
5.872 |
T P |
| 2r62_A |
249 |
388 |
142 |
2.38 |
10.56 |
24.064 |
5.722 |
T P |
| 2rko_A |
280 |
388 |
143 |
2.44 |
11.89 |
23.861 |
5.629 |
T P |
| 1njg_A |
240 |
388 |
142 |
2.48 |
11.97 |
23.580 |
5.506 |
T P |
| 1sxj_C |
322 |
388 |
134 |
2.24 |
11.19 |
23.491 |
5.730 |
T P |
| 3f8t_A |
459 |
388 |
132 |
2.21 |
11.36 |
23.249 |
5.707 |
T P |
| 1sxj_B |
316 |
388 |
136 |
2.22 |
11.03 |
23.247 |
5.850 |
T P |
| 1lv7_A |
251 |
388 |
135 |
2.45 |
10.37 |
22.350 |
5.303 |
T P |
| 1d2n_A |
246 |
388 |
122 |
2.38 |
13.93 |
21.171 |
4.923 |
T P |
| 3f9v_A |
595 |
388 |
130 |
2.48 |
7.69 |
21.165 |
5.040 |
T P |
| 1qzm_A |
94 |
388 |
69 |
2.31 |
7.25 |
11.892 |
2.867 |
T P |
| 1x37_A |
95 |
388 |
63 |
2.43 |
3.17 |
11.248 |
2.495 |
T P |
| 2qjw_A |
176 |
388 |
61 |
2.65 |
4.92 |
9.686 |
2.220 |
T P |
| 3fh2_A |
146 |
388 |
48 |
2.58 |
8.33 |
8.505 |
1.794 |
T P |
| 1khy_D |
140 |
388 |
43 |
2.76 |
2.33 |
7.827 |
1.503 |
T P |
| 1xhk_B |
187 |
388 |
38 |
2.56 |
7.89 |
7.105 |
1.427 |
T P |
| 1z0e_A |
195 |
388 |
40 |
2.80 |
17.50 |
6.761 |
1.380 |
T P |
| 1rre_E |
191 |
388 |
39 |
2.52 |
5.13 |
6.730 |
1.489 |
T P |
| 1z0b_A |
203 |
388 |
34 |
2.87 |
0.00 |
6.003 |
1.145 |
T P |
| 1z0c_A |
200 |
388 |
33 |
2.67 |
12.12 |
5.672 |
1.192 |
T P |
| 1z0w_A |
203 |
388 |
27 |
2.48 |
0.00 |
5.429 |
1.048 |
T P |
| 1rr9_E |
191 |
388 |
24 |
2.11 |
0.00 |
4.997 |
1.088 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]