LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_79.5wLII_11067_27
Total number of 3D structures: 127
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
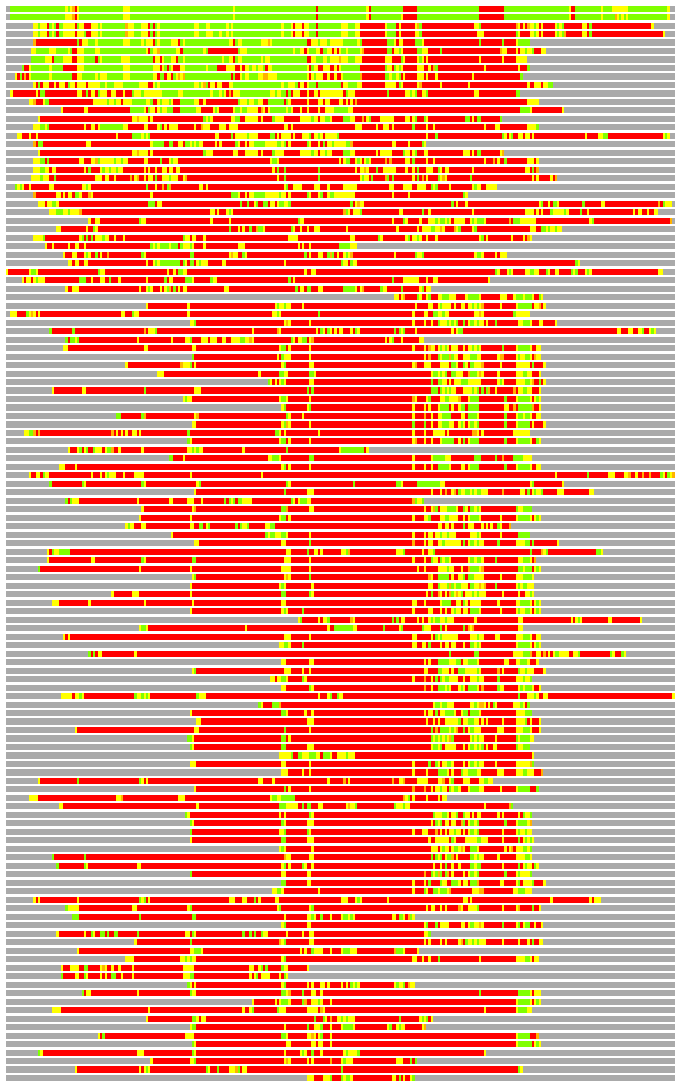
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1tv8_A |
327 |
291 |
265 |
1.01 |
19.25 |
88.660 |
23.971 |
T P |
| 2fb2_A |
327 |
291 |
265 |
1.05 |
19.25 |
88.509 |
23.026 |
T P |
| 3ciw_A |
346 |
291 |
160 |
2.16 |
7.50 |
42.048 |
7.069 |
T P |
| 3cix_A |
346 |
291 |
160 |
2.16 |
7.50 |
41.926 |
7.075 |
T P |
| 3can_A |
161 |
291 |
137 |
1.93 |
13.87 |
40.232 |
6.746 |
T P |
| 2a5h_B |
410 |
291 |
148 |
2.01 |
11.49 |
40.100 |
7.020 |
T P |
| 3c8f_A |
245 |
291 |
150 |
1.95 |
11.33 |
40.017 |
7.308 |
T P |
| 2z2u_A |
279 |
291 |
136 |
2.02 |
16.18 |
39.333 |
6.406 |
T P |
| 2yx0_A |
324 |
291 |
148 |
2.04 |
18.24 |
38.152 |
6.918 |
T P |
| 1r30_B |
313 |
291 |
143 |
2.27 |
11.89 |
35.443 |
6.028 |
T P |
| 2qgq_A |
272 |
291 |
115 |
2.48 |
10.43 |
26.655 |
4.466 |
T P |
| 1x1o_A |
277 |
291 |
82 |
2.59 |
3.66 |
18.350 |
3.051 |
T P |
| 1fc9_A |
386 |
291 |
77 |
2.27 |
3.90 |
18.268 |
3.242 |
T P |
| 1jxm_A |
264 |
291 |
68 |
2.63 |
8.82 |
14.826 |
2.494 |
T P |
| 1qha_A |
903 |
291 |
66 |
2.78 |
12.12 |
14.800 |
2.291 |
T P |
| 2b0t_A |
735 |
291 |
67 |
2.74 |
2.99 |
14.796 |
2.362 |
T P |
| 1ecx_B |
365 |
291 |
65 |
2.45 |
12.31 |
14.513 |
2.548 |
T P |
| 1kjw_A |
292 |
291 |
66 |
2.80 |
7.58 |
14.065 |
2.275 |
T P |
| 1bg3_A |
902 |
291 |
60 |
2.73 |
13.33 |
13.924 |
2.120 |
T P |
| 1cza_N |
898 |
291 |
61 |
2.85 |
1.64 |
13.712 |
2.069 |
T P |
| 1dgk_N |
898 |
291 |
55 |
2.89 |
7.27 |
12.827 |
1.841 |
T P |
| 1fc6_A |
386 |
291 |
58 |
2.95 |
1.72 |
12.533 |
1.900 |
T P |
| 1eg5_B |
365 |
291 |
54 |
2.71 |
9.26 |
11.715 |
1.920 |
T P |
| 1ky9_B |
396 |
291 |
49 |
2.58 |
4.08 |
11.699 |
1.827 |
T P |
| 3cs0_A |
392 |
291 |
54 |
2.76 |
7.41 |
11.665 |
1.886 |
T P |
| 1soz_A |
281 |
291 |
51 |
2.64 |
5.88 |
11.546 |
1.863 |
T P |
| 1lcy_A |
296 |
291 |
50 |
2.62 |
6.00 |
11.472 |
1.841 |
T P |
| 1hkb_A |
899 |
291 |
46 |
2.87 |
10.87 |
10.143 |
1.549 |
T P |
| 2ytw_A |
118 |
291 |
39 |
2.58 |
5.13 |
9.866 |
1.453 |
T P |
| 2qt5_A |
194 |
291 |
44 |
2.83 |
6.82 |
9.783 |
1.503 |
T P |
| 2pzd_A |
106 |
291 |
39 |
2.35 |
12.82 |
9.727 |
1.593 |
T P |
| 1itw_A |
740 |
291 |
40 |
2.61 |
5.00 |
9.447 |
1.475 |
T P |
| 1sot_B |
294 |
291 |
41 |
2.68 |
7.32 |
9.294 |
1.477 |
T P |
| 1p1d_A |
196 |
291 |
41 |
2.64 |
9.76 |
9.199 |
1.498 |
T P |
| 2zpl_B |
94 |
291 |
37 |
2.28 |
8.11 |
9.180 |
1.556 |
T P |
| 1d5g_A |
96 |
291 |
41 |
2.75 |
2.44 |
9.170 |
1.440 |
T P |
| 1n99_B |
163 |
291 |
40 |
2.68 |
5.00 |
9.007 |
1.440 |
T P |
| 2ozf_A |
92 |
291 |
38 |
2.50 |
10.53 |
8.991 |
1.463 |
T P |
| 1z87_A |
263 |
291 |
40 |
2.80 |
10.00 |
8.912 |
1.380 |
T P |
| 1tp5_A |
115 |
291 |
42 |
2.77 |
7.14 |
8.884 |
1.465 |
T P |
| 2jin_A |
102 |
291 |
41 |
2.69 |
4.88 |
8.847 |
1.468 |
T P |
| 1ozi_A |
99 |
291 |
39 |
2.75 |
7.69 |
8.791 |
1.370 |
T P |
| 2eno_A |
120 |
291 |
40 |
2.75 |
7.50 |
8.788 |
1.403 |
T P |
| 1uju_A |
111 |
291 |
39 |
2.59 |
5.13 |
8.725 |
1.448 |
T P |
| 1p1e_A |
101 |
291 |
39 |
2.77 |
7.69 |
8.697 |
1.360 |
T P |
| 2eej_A |
96 |
291 |
38 |
2.59 |
5.26 |
8.693 |
1.415 |
T P |
| 2byg_A |
98 |
291 |
38 |
2.46 |
7.89 |
8.609 |
1.482 |
T P |
| 2dc2_A |
87 |
291 |
32 |
2.30 |
6.25 |
8.605 |
1.333 |
T P |
| 1v5q_A |
122 |
291 |
35 |
2.44 |
2.86 |
8.544 |
1.378 |
T P |
| 1n7e_A |
95 |
291 |
34 |
2.38 |
8.82 |
8.478 |
1.370 |
T P |
| 1w9e_A |
164 |
291 |
38 |
3.06 |
2.63 |
8.462 |
1.203 |
T P |
| 2eaq_A |
89 |
291 |
34 |
2.37 |
11.76 |
8.430 |
1.374 |
T P |
| 2joa_A |
105 |
291 |
36 |
2.41 |
11.11 |
8.424 |
1.436 |
T P |
| 1uez_A |
101 |
291 |
34 |
2.29 |
11.76 |
8.400 |
1.422 |
T P |
| 1vb7_A |
94 |
291 |
34 |
2.44 |
8.82 |
8.314 |
1.341 |
T P |
| 1b8q_A |
127 |
291 |
36 |
3.00 |
5.56 |
8.310 |
1.161 |
T P |
| 2p3w_A |
110 |
291 |
28 |
2.00 |
10.71 |
8.254 |
1.332 |
T P |
| 2edz_A |
114 |
291 |
37 |
2.93 |
8.11 |
8.250 |
1.220 |
T P |
| 1be9_A |
115 |
291 |
35 |
2.41 |
8.57 |
8.247 |
1.394 |
T P |
| 1qav_B |
115 |
291 |
34 |
2.35 |
14.71 |
8.246 |
1.386 |
T P |
| 2fe5_A |
94 |
291 |
36 |
2.50 |
5.56 |
8.229 |
1.383 |
T P |
| 1te0_A |
318 |
291 |
36 |
2.72 |
5.56 |
8.227 |
1.277 |
T P |
| 2fne_C |
104 |
291 |
36 |
2.53 |
8.33 |
8.227 |
1.368 |
T P |
| 1gq4_A |
90 |
291 |
36 |
2.63 |
5.56 |
8.167 |
1.320 |
T P |
| 1uf1_A |
128 |
291 |
34 |
2.62 |
11.76 |
8.157 |
1.250 |
T P |
| 1whd_A |
100 |
291 |
36 |
2.42 |
5.56 |
8.128 |
1.430 |
T P |
| 2omj_A |
89 |
291 |
34 |
2.57 |
5.88 |
8.019 |
1.272 |
T P |
| 1qav_A |
90 |
291 |
33 |
2.46 |
9.09 |
8.014 |
1.289 |
T P |
| 1iu0_A |
91 |
291 |
36 |
2.70 |
11.11 |
7.956 |
1.287 |
T P |
| 1wha_A |
105 |
291 |
35 |
2.80 |
11.43 |
7.904 |
1.209 |
T P |
| 2eeh_A |
100 |
291 |
36 |
2.83 |
8.33 |
7.858 |
1.228 |
T P |
| 2g2l_A |
90 |
291 |
32 |
2.65 |
6.25 |
7.858 |
1.164 |
T P |
| 2hga_A |
103 |
291 |
35 |
2.65 |
8.57 |
7.852 |
1.273 |
T P |
| 1wi2_A |
104 |
291 |
31 |
2.49 |
9.68 |
7.848 |
1.195 |
T P |
| 2dkr_A |
93 |
291 |
36 |
2.76 |
11.11 |
7.839 |
1.259 |
T P |
| 2awx_A |
92 |
291 |
32 |
2.50 |
6.25 |
7.839 |
1.229 |
T P |
| 2yuy_A |
126 |
291 |
34 |
2.63 |
11.76 |
7.831 |
1.244 |
T P |
| 1rgr_A |
93 |
291 |
32 |
2.37 |
9.38 |
7.826 |
1.298 |
T P |
| 2he2_A |
102 |
291 |
35 |
2.65 |
5.71 |
7.789 |
1.271 |
T P |
| 1gm1_A |
94 |
291 |
36 |
2.68 |
2.78 |
7.784 |
1.296 |
T P |
| 2pdz_A |
86 |
291 |
32 |
2.39 |
9.38 |
7.755 |
1.283 |
T P |
| 2qbw_A |
189 |
291 |
35 |
2.70 |
5.71 |
7.749 |
1.251 |
T P |
| 2he4_A |
90 |
291 |
31 |
2.61 |
3.23 |
7.747 |
1.143 |
T P |
| 1qau_A |
112 |
291 |
32 |
2.33 |
15.62 |
7.740 |
1.319 |
T P |
| 1i92_A |
91 |
291 |
32 |
2.92 |
0.00 |
7.732 |
1.058 |
T P |
| 1qlc_A |
95 |
291 |
33 |
2.63 |
3.03 |
7.689 |
1.208 |
T P |
| 2pkt_A |
91 |
291 |
31 |
2.39 |
0.00 |
7.648 |
1.243 |
T P |
| 2iwq_A |
92 |
291 |
34 |
2.54 |
8.82 |
7.613 |
1.286 |
T P |
| 2egk_B |
90 |
291 |
29 |
2.45 |
3.45 |
7.600 |
1.138 |
T P |
| 2f5y_A |
82 |
291 |
32 |
2.58 |
3.12 |
7.543 |
1.195 |
T P |
| 2vrf_A |
95 |
291 |
34 |
2.62 |
8.82 |
7.533 |
1.250 |
T P |
| 1wh1_A |
124 |
291 |
34 |
2.89 |
2.94 |
7.532 |
1.139 |
T P |
| 1kef_A |
93 |
291 |
32 |
2.67 |
6.25 |
7.456 |
1.154 |
T P |
| 1x5r_A |
112 |
291 |
29 |
2.75 |
10.34 |
7.433 |
1.018 |
T P |
| 1q7x_A |
108 |
291 |
31 |
2.66 |
3.23 |
7.385 |
1.125 |
T P |
| 1ihj_B |
95 |
291 |
29 |
2.70 |
10.34 |
7.373 |
1.035 |
T P |
| 2vz5_A |
111 |
291 |
30 |
2.45 |
3.33 |
7.358 |
1.176 |
T P |
| 2awu_A |
95 |
291 |
32 |
2.65 |
6.25 |
7.347 |
1.163 |
T P |
| 3dj3_B |
101 |
291 |
32 |
2.74 |
9.38 |
7.338 |
1.128 |
T P |
| 2w4f_A |
95 |
291 |
31 |
2.58 |
3.23 |
7.293 |
1.155 |
T P |
| 1uew_A |
114 |
291 |
30 |
2.55 |
6.67 |
7.212 |
1.134 |
T P |
| 1x5n_A |
114 |
291 |
29 |
2.59 |
6.90 |
7.181 |
1.080 |
T P |
| 2ocs_A |
85 |
291 |
28 |
2.39 |
7.14 |
7.159 |
1.124 |
T P |
| 2oqs_A |
92 |
291 |
30 |
2.47 |
6.67 |
7.109 |
1.168 |
T P |
| 2jil_B |
97 |
291 |
30 |
2.62 |
6.67 |
7.059 |
1.103 |
T P |
| 1v5l_A |
103 |
291 |
31 |
2.98 |
3.23 |
6.864 |
1.007 |
T P |
| 2dlu_A |
111 |
291 |
31 |
2.96 |
0.00 |
6.838 |
1.012 |
T P |
| 1uhp_A |
107 |
291 |
27 |
2.39 |
3.70 |
6.810 |
1.085 |
T P |
| 1z86_A |
87 |
291 |
28 |
2.68 |
10.71 |
6.732 |
1.007 |
T P |
| 2d90_A |
102 |
291 |
27 |
2.36 |
11.11 |
6.551 |
1.096 |
T P |
| 2vsp_A |
83 |
291 |
28 |
2.80 |
0.00 |
6.447 |
0.965 |
T P |
| 3dj1_A |
115 |
291 |
28 |
2.59 |
10.71 |
6.381 |
1.040 |
T P |
| 1u3b_A |
185 |
291 |
28 |
2.79 |
3.57 |
6.292 |
0.969 |
T P |
| 2yt8_A |
94 |
291 |
27 |
2.73 |
3.70 |
6.240 |
0.955 |
T P |
| 1gq5_A |
91 |
291 |
26 |
2.62 |
3.85 |
6.222 |
0.956 |
T P |
| 2v90_A |
95 |
291 |
24 |
2.77 |
12.50 |
6.065 |
0.835 |
T P |
| 1uit_A |
117 |
291 |
25 |
2.50 |
0.00 |
5.999 |
0.960 |
T P |
| 1vj6_A |
95 |
291 |
25 |
2.55 |
8.00 |
5.974 |
0.945 |
T P |
| 1um7_A |
113 |
291 |
27 |
2.80 |
7.41 |
5.774 |
0.930 |
T P |
| 2ego_B |
90 |
291 |
24 |
2.58 |
4.17 |
5.604 |
0.897 |
T P |
| 1g9o_A |
91 |
291 |
24 |
2.69 |
4.17 |
5.579 |
0.860 |
T P |
| 1x5q_A |
110 |
291 |
23 |
2.74 |
4.35 |
5.291 |
0.809 |
T P |
| 2fcf_A |
89 |
291 |
19 |
2.57 |
10.53 |
5.070 |
0.712 |
T P |
| 2dls_A |
93 |
291 |
21 |
2.51 |
0.00 |
5.027 |
0.805 |
T P |
| 2dmz_A |
129 |
291 |
20 |
2.64 |
0.00 |
4.962 |
0.730 |
T P |
| 2zpm_A |
86 |
291 |
18 |
2.67 |
0.00 |
4.671 |
0.651 |
T P |
| 2i0l_A |
83 |
291 |
18 |
2.80 |
5.56 |
4.019 |
0.621 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]