LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_85.5wLII_11067_43
Total number of 3D structures: 63
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
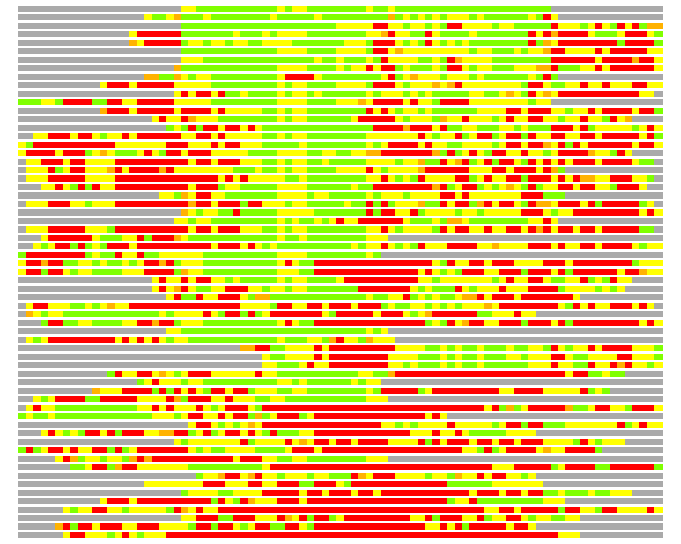
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1deq_A |
180 |
87 |
50 |
1.27 |
22.00 |
55.878 |
3.658 |
T P |
| 1jk0_A |
334 |
87 |
55 |
1.99 |
5.45 |
55.551 |
2.635 |
T P |
| 2dq3_A |
425 |
87 |
57 |
2.16 |
7.02 |
52.275 |
2.520 |
T P |
| 1bf5_A |
545 |
87 |
55 |
2.07 |
9.09 |
49.650 |
2.539 |
T P |
| 1yvl_B |
653 |
87 |
50 |
2.04 |
12.00 |
48.451 |
2.340 |
T P |
| 1wle_B |
469 |
87 |
55 |
2.33 |
16.36 |
47.999 |
2.260 |
T P |
| 2dq0_A |
447 |
87 |
51 |
2.16 |
5.88 |
46.094 |
2.256 |
T P |
| 1ses_A |
421 |
87 |
53 |
2.23 |
11.32 |
44.696 |
2.277 |
T P |
| 1smq_A |
329 |
87 |
50 |
2.15 |
6.00 |
44.675 |
2.226 |
T P |
| 1cii_A |
602 |
87 |
58 |
2.52 |
6.90 |
44.409 |
2.217 |
T P |
| 1jch_A |
468 |
87 |
49 |
2.35 |
14.29 |
43.291 |
1.999 |
T P |
| 1rrj_A |
565 |
87 |
52 |
2.52 |
7.69 |
43.174 |
1.985 |
T P |
| 2b5u_A |
470 |
87 |
49 |
2.49 |
10.20 |
42.300 |
1.890 |
T P |
| 1n52_A |
732 |
87 |
52 |
2.60 |
9.62 |
42.042 |
1.929 |
T P |
| 3ffz_A |
1246 |
87 |
48 |
2.22 |
0.00 |
41.192 |
2.071 |
T P |
| 2hko_A |
647 |
87 |
53 |
2.64 |
9.43 |
41.091 |
1.935 |
T P |
| 2uv8_A |
1613 |
87 |
51 |
2.51 |
7.84 |
41.032 |
1.952 |
T P |
| 2vkz_A |
1614 |
87 |
53 |
2.76 |
11.32 |
40.827 |
1.850 |
T P |
| 2h94_A |
647 |
87 |
50 |
2.67 |
10.00 |
40.634 |
1.803 |
T P |
| 1nt2_B |
236 |
87 |
51 |
2.65 |
7.84 |
40.497 |
1.856 |
T P |
| 2z3y_A |
643 |
87 |
49 |
2.59 |
10.20 |
39.726 |
1.819 |
T P |
| 2pff_A |
1683 |
87 |
47 |
2.39 |
8.51 |
39.652 |
1.889 |
T P |
| 1lrz_A |
400 |
87 |
47 |
2.46 |
6.38 |
39.618 |
1.837 |
T P |
| 2iw5_A |
666 |
87 |
47 |
2.56 |
8.51 |
39.535 |
1.769 |
T P |
| 1qvr_A |
803 |
87 |
47 |
2.42 |
2.13 |
38.889 |
1.868 |
T P |
| 1zvu_A |
685 |
87 |
44 |
2.34 |
9.09 |
38.624 |
1.807 |
T P |
| 2dw4_A |
634 |
87 |
47 |
2.49 |
14.89 |
38.259 |
1.814 |
T P |
| 1ha0_A |
494 |
87 |
37 |
1.70 |
2.70 |
37.849 |
2.057 |
T P |
| 2v1d_A |
666 |
87 |
49 |
2.56 |
10.20 |
37.654 |
1.841 |
T P |
| 2zuo_A |
812 |
87 |
39 |
2.05 |
12.82 |
37.539 |
1.812 |
T P |
| 1lpq_A |
557 |
87 |
46 |
2.56 |
13.04 |
37.167 |
1.732 |
T P |
| 1seu_A |
565 |
87 |
46 |
2.38 |
10.87 |
36.948 |
1.856 |
T P |
| 1h2v_C |
722 |
87 |
46 |
2.47 |
6.52 |
36.759 |
1.788 |
T P |
| 1h6k_C |
733 |
87 |
46 |
2.42 |
15.22 |
36.680 |
1.825 |
T P |
| 1bg1_A |
559 |
87 |
39 |
2.38 |
7.69 |
36.139 |
1.571 |
T P |
| 2rd0_B |
139 |
87 |
44 |
2.82 |
2.27 |
35.732 |
1.508 |
T P |
| 1pix_A |
586 |
87 |
43 |
2.51 |
6.98 |
35.177 |
1.646 |
T P |
| 1k4t_A |
565 |
87 |
42 |
2.34 |
11.90 |
34.888 |
1.724 |
T P |
| 2cly_A |
105 |
87 |
30 |
1.48 |
0.00 |
34.029 |
1.895 |
T P |
| 2qzv_A |
749 |
87 |
37 |
2.25 |
13.51 |
33.866 |
1.575 |
T P |
| 3eyk_B |
172 |
87 |
39 |
2.48 |
7.69 |
33.187 |
1.512 |
T P |
| 3eym_B |
172 |
87 |
41 |
2.40 |
7.32 |
33.176 |
1.640 |
T P |
| 1mqm_B |
172 |
87 |
41 |
2.48 |
9.76 |
32.916 |
1.591 |
T P |
| 1f5n_A |
570 |
87 |
38 |
2.39 |
7.89 |
32.603 |
1.528 |
T P |
| 2iqi_F |
175 |
87 |
32 |
2.09 |
3.12 |
32.357 |
1.461 |
T P |
| 1yuw_A |
554 |
87 |
40 |
2.39 |
5.00 |
32.203 |
1.604 |
T P |
| 1ii8_A |
195 |
87 |
34 |
2.17 |
2.94 |
31.879 |
1.495 |
T P |
| 2bde_A |
458 |
87 |
38 |
2.60 |
2.63 |
31.451 |
1.406 |
T P |
| 2ve7_B |
303 |
87 |
34 |
2.24 |
17.65 |
30.397 |
1.450 |
T P |
| 2viu_B |
175 |
87 |
39 |
2.72 |
2.56 |
29.585 |
1.382 |
T P |
| 2zux_A |
582 |
87 |
38 |
2.44 |
13.16 |
29.539 |
1.496 |
T P |
| 1y1u_A |
544 |
87 |
38 |
2.88 |
5.26 |
29.120 |
1.274 |
T P |
| 1giq_A |
411 |
87 |
34 |
2.42 |
8.82 |
27.771 |
1.349 |
T P |
| 1c1g_A |
284 |
87 |
29 |
2.45 |
0.00 |
27.354 |
1.136 |
T P |
| 1oqy_A |
363 |
87 |
31 |
2.30 |
9.68 |
27.319 |
1.290 |
T P |
| 1ei3_C |
397 |
87 |
36 |
2.88 |
5.56 |
26.071 |
1.208 |
T P |
| 2tec_E |
279 |
87 |
30 |
2.58 |
10.00 |
25.289 |
1.118 |
T P |
| 3cwg_B |
507 |
87 |
30 |
2.62 |
10.00 |
24.647 |
1.104 |
T P |
| 1thm_A |
279 |
87 |
25 |
2.46 |
0.00 |
22.774 |
0.978 |
T P |
| 1m1j_C |
390 |
87 |
29 |
2.96 |
6.90 |
22.110 |
0.948 |
T P |
| 1arb_A |
263 |
87 |
28 |
2.79 |
0.00 |
20.496 |
0.969 |
T P |
| 1nkg_A |
508 |
87 |
24 |
2.63 |
4.17 |
19.610 |
0.881 |
T P |
| 2wb7_A |
525 |
87 |
14 |
2.64 |
0.00 |
12.375 |
0.511 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]