LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_88.5wLII_11068_5
Total number of 3D structures: 81
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
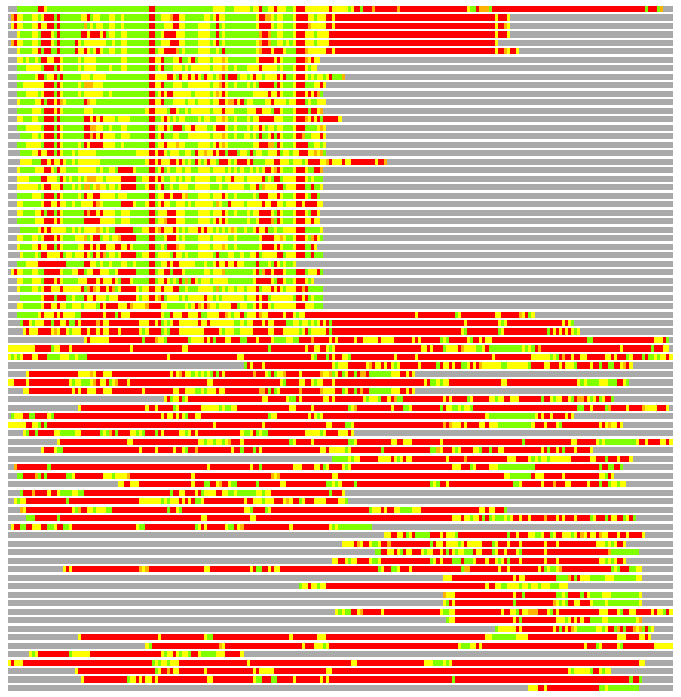
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 2eyq_A |
1146 |
217 |
113 |
2.11 |
12.39 |
46.000 |
5.123 |
T P |
| 1t5l_A |
595 |
217 |
89 |
2.11 |
11.24 |
32.603 |
4.020 |
T P |
| 2d7d_A |
621 |
217 |
89 |
2.28 |
10.11 |
30.571 |
3.742 |
T P |
| 2fdc_B |
585 |
217 |
82 |
2.06 |
8.54 |
30.041 |
3.798 |
T P |
| 1d9z_A |
590 |
217 |
88 |
2.13 |
10.23 |
29.705 |
3.952 |
T P |
| 1d9x_A |
590 |
217 |
83 |
2.20 |
9.64 |
29.357 |
3.601 |
T P |
| 3eaq_B |
209 |
217 |
80 |
2.10 |
7.50 |
28.978 |
3.640 |
T P |
| 1hv8_A |
363 |
217 |
87 |
2.30 |
9.20 |
27.931 |
3.631 |
T P |
| 1gm5_A |
729 |
217 |
84 |
2.26 |
13.10 |
27.789 |
3.558 |
T P |
| 2i4i_A |
408 |
217 |
83 |
2.26 |
7.23 |
27.441 |
3.512 |
T P |
| 1t5i_A |
159 |
217 |
83 |
2.25 |
4.82 |
27.424 |
3.530 |
T P |
| 3ews_A |
416 |
217 |
85 |
2.28 |
14.12 |
27.414 |
3.571 |
T P |
| 1fuk_A |
157 |
217 |
81 |
2.18 |
8.64 |
26.923 |
3.559 |
T P |
| 1oyw_A |
516 |
217 |
80 |
2.24 |
12.50 |
26.487 |
3.423 |
T P |
| 2jgn_B |
161 |
217 |
81 |
2.22 |
8.64 |
26.407 |
3.497 |
T P |
| 2p6n_A |
160 |
217 |
83 |
2.24 |
10.84 |
26.343 |
3.553 |
T P |
| 2db3_A |
420 |
217 |
80 |
2.21 |
5.00 |
26.168 |
3.458 |
T P |
| 2vso_A |
366 |
217 |
80 |
2.40 |
11.25 |
25.679 |
3.199 |
T P |
| 2p6r_A |
683 |
217 |
83 |
2.52 |
12.05 |
25.361 |
3.169 |
T P |
| 1wp9_A |
479 |
217 |
81 |
2.33 |
9.88 |
24.603 |
3.327 |
T P |
| 2z0m_A |
331 |
217 |
82 |
2.48 |
9.76 |
24.500 |
3.173 |
T P |
| 2hyi_C |
392 |
217 |
82 |
2.50 |
7.32 |
24.392 |
3.155 |
T P |
| 1xtj_A |
374 |
217 |
75 |
2.25 |
5.33 |
24.312 |
3.188 |
T P |
| 3eiq_D |
371 |
217 |
79 |
2.42 |
8.86 |
24.031 |
3.129 |
T P |
| 3fht_A |
392 |
217 |
75 |
2.28 |
13.33 |
23.870 |
3.156 |
T P |
| 3g0h_A |
408 |
217 |
74 |
2.28 |
13.51 |
23.843 |
3.106 |
T P |
| 1xtk_A |
382 |
217 |
80 |
2.49 |
7.50 |
23.498 |
3.092 |
T P |
| 2rb4_A |
164 |
217 |
76 |
2.37 |
6.58 |
23.482 |
3.082 |
T P |
| 2g2j_A |
158 |
217 |
72 |
2.27 |
9.72 |
23.392 |
3.036 |
T P |
| 2j0s_A |
391 |
217 |
77 |
2.44 |
6.49 |
23.316 |
3.034 |
T P |
| 2fzl_A |
197 |
217 |
70 |
2.21 |
8.57 |
22.996 |
3.024 |
T P |
| 1xti_A |
381 |
217 |
69 |
2.13 |
5.80 |
22.386 |
3.096 |
T P |
| 2hjv_A |
158 |
217 |
70 |
2.32 |
5.71 |
22.343 |
2.898 |
T P |
| 1s2m_A |
377 |
217 |
76 |
2.51 |
9.21 |
22.268 |
2.909 |
T P |
| 2zu6_C |
351 |
217 |
75 |
2.50 |
9.33 |
21.963 |
2.884 |
T P |
| 1fuu_B |
380 |
217 |
77 |
2.63 |
6.49 |
21.806 |
2.818 |
T P |
| 2j0u_A |
366 |
217 |
64 |
2.57 |
1.56 |
19.771 |
2.394 |
T P |
| 2hxy_A |
376 |
217 |
68 |
2.73 |
10.29 |
19.599 |
2.403 |
T P |
| 2j0u_B |
347 |
217 |
69 |
2.80 |
8.70 |
19.528 |
2.380 |
T P |
| 2ahu_A |
515 |
217 |
62 |
2.60 |
11.29 |
18.756 |
2.299 |
T P |
| 1qvr_A |
803 |
217 |
59 |
2.58 |
13.56 |
18.462 |
2.199 |
T P |
| 1wz2_A |
948 |
217 |
59 |
2.67 |
6.78 |
18.132 |
2.132 |
T P |
| 1yuw_A |
554 |
217 |
56 |
2.77 |
1.79 |
17.711 |
1.948 |
T P |
| 2z3y_A |
643 |
217 |
55 |
2.81 |
5.45 |
16.651 |
1.889 |
T P |
| 2fwr_A |
434 |
217 |
53 |
2.53 |
7.55 |
16.388 |
2.015 |
T P |
| 2h94_A |
647 |
217 |
52 |
2.81 |
7.69 |
15.891 |
1.790 |
T P |
| 2iw5_A |
666 |
217 |
52 |
2.73 |
3.85 |
15.746 |
1.838 |
T P |
| 3cwg_B |
507 |
217 |
49 |
2.74 |
4.08 |
15.128 |
1.727 |
T P |
| 1lrz_A |
400 |
217 |
44 |
2.37 |
6.82 |
15.101 |
1.784 |
T P |
| 1pyy_A |
607 |
217 |
49 |
2.73 |
4.08 |
14.997 |
1.730 |
T P |
| 1znn_A |
245 |
217 |
48 |
2.61 |
12.50 |
14.922 |
1.769 |
T P |
| 1bg1_A |
559 |
217 |
48 |
2.67 |
6.25 |
14.756 |
1.732 |
T P |
| 1ei3_C |
397 |
217 |
43 |
2.69 |
4.65 |
13.992 |
1.542 |
T P |
| 2hko_A |
647 |
217 |
42 |
2.70 |
9.52 |
13.680 |
1.502 |
T P |
| 1rp5_A |
687 |
217 |
39 |
2.32 |
0.00 |
13.322 |
1.613 |
T P |
| 1f5n_A |
570 |
217 |
40 |
2.55 |
5.00 |
13.086 |
1.511 |
T P |
| 1ha0_A |
494 |
217 |
41 |
2.62 |
2.44 |
13.060 |
1.510 |
T P |
| 1cii_A |
602 |
217 |
39 |
2.40 |
12.82 |
13.052 |
1.562 |
T P |
| 2iss_B |
285 |
217 |
43 |
2.83 |
6.98 |
12.699 |
1.468 |
T P |
| 1m1j_C |
390 |
217 |
40 |
2.66 |
5.00 |
12.680 |
1.450 |
T P |
| 1pmd_A |
675 |
217 |
39 |
2.67 |
5.13 |
12.650 |
1.407 |
T P |
| 1mqm_B |
172 |
217 |
38 |
2.24 |
10.53 |
12.510 |
1.623 |
T P |
| 2inr_A |
442 |
217 |
40 |
2.65 |
12.50 |
12.398 |
1.454 |
T P |
| 3eyk_B |
172 |
217 |
39 |
2.77 |
2.56 |
12.142 |
1.361 |
T P |
| 3eym_B |
172 |
217 |
39 |
2.57 |
0.00 |
11.871 |
1.463 |
T P |
| 5hmg_B |
175 |
217 |
35 |
2.50 |
2.86 |
11.437 |
1.348 |
T P |
| 2v1d_A |
666 |
217 |
38 |
2.76 |
0.00 |
11.252 |
1.330 |
T P |
| 1qu7_A |
227 |
217 |
32 |
2.28 |
6.25 |
10.995 |
1.343 |
T P |
| 1deq_A |
180 |
217 |
33 |
2.51 |
6.06 |
10.885 |
1.262 |
T P |
| 2ch7_A |
309 |
217 |
29 |
2.28 |
0.00 |
10.801 |
1.220 |
T P |
| 2ch7_B |
307 |
217 |
28 |
2.18 |
7.14 |
10.732 |
1.230 |
T P |
| 2qdl_A |
154 |
217 |
37 |
2.84 |
5.41 |
10.520 |
1.257 |
T P |
| 2rd0_B |
139 |
217 |
29 |
2.26 |
13.79 |
10.444 |
1.229 |
T P |
| 2v0o_A |
273 |
217 |
32 |
2.61 |
0.00 |
10.293 |
1.181 |
T P |
| 1kmi_Z |
177 |
217 |
29 |
2.51 |
10.34 |
10.156 |
1.110 |
T P |
| 2viu_B |
175 |
217 |
28 |
2.72 |
3.57 |
9.147 |
0.994 |
T P |
| 1c07_A |
95 |
217 |
29 |
2.60 |
3.45 |
8.865 |
1.074 |
T P |
| 1c1g_A |
284 |
217 |
27 |
2.63 |
7.41 |
8.216 |
0.988 |
T P |
| 1qu1_F |
155 |
217 |
27 |
2.97 |
7.41 |
7.985 |
0.880 |
T P |
| 2dw4_A |
634 |
217 |
21 |
2.51 |
4.76 |
7.374 |
0.803 |
T P |
| 2pl9_D |
19 |
217 |
17 |
1.71 |
0.00 |
7.337 |
0.938 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]