LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_92.5wLII_11068_40
Total number of 3D structures: 39
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
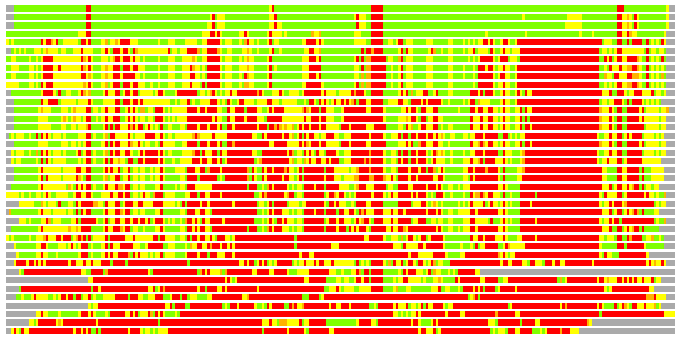
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1t70_A |
255 |
269 |
253 |
0.38 |
42.29 |
93.737 |
52.988 |
T P |
| 2cv9_A |
252 |
269 |
251 |
1.18 |
40.64 |
90.100 |
19.641 |
T P |
| 2z06_A |
252 |
269 |
251 |
1.21 |
41.04 |
89.902 |
19.210 |
T P |
| 1t71_A |
281 |
269 |
251 |
1.38 |
32.27 |
88.823 |
16.910 |
T P |
| 1hp1_A |
516 |
269 |
188 |
2.23 |
14.89 |
47.153 |
8.082 |
T P |
| 2z1a_A |
507 |
269 |
196 |
2.37 |
16.33 |
46.926 |
7.940 |
T P |
| 1oid_A |
525 |
269 |
185 |
2.28 |
15.68 |
45.290 |
7.776 |
T P |
| 1hpu_A |
525 |
269 |
187 |
2.32 |
15.51 |
45.166 |
7.723 |
T P |
| 1oi8_A |
525 |
269 |
186 |
2.31 |
15.59 |
45.155 |
7.708 |
T P |
| 1ush_A |
515 |
269 |
186 |
2.34 |
15.59 |
44.932 |
7.608 |
T P |
| 2q8u_A |
308 |
269 |
161 |
2.40 |
13.04 |
37.828 |
6.439 |
T P |
| 3c9f_B |
540 |
269 |
166 |
2.52 |
13.25 |
37.591 |
6.325 |
T P |
| 3dsc_A |
333 |
269 |
158 |
2.45 |
9.49 |
36.196 |
6.186 |
T P |
| 3dsd_A |
333 |
269 |
154 |
2.47 |
8.44 |
35.366 |
5.989 |
T P |
| 1ii7_A |
333 |
269 |
152 |
2.47 |
11.18 |
34.956 |
5.912 |
T P |
| 1ute_A |
302 |
269 |
153 |
2.61 |
20.26 |
34.870 |
5.652 |
T P |
| 1s8e_A |
333 |
269 |
150 |
2.44 |
11.33 |
34.502 |
5.907 |
T P |
| 1qhw_A |
300 |
269 |
145 |
2.51 |
20.69 |
34.381 |
5.558 |
T P |
| 1qfc_A |
287 |
269 |
144 |
2.60 |
21.53 |
33.750 |
5.342 |
T P |
| 3d03_A |
274 |
269 |
141 |
2.52 |
13.48 |
33.597 |
5.372 |
T P |
| 2dxn_A |
271 |
269 |
137 |
2.41 |
15.33 |
33.359 |
5.465 |
T P |
| 2hyp_A |
230 |
269 |
140 |
2.42 |
16.43 |
33.058 |
5.551 |
T P |
| 1xzw_B |
426 |
269 |
138 |
2.59 |
14.49 |
32.754 |
5.127 |
T P |
| 1kbp_A |
424 |
269 |
134 |
2.50 |
17.16 |
32.513 |
5.164 |
T P |
| 2hyo_A |
227 |
269 |
138 |
2.43 |
15.94 |
32.471 |
5.449 |
T P |
| 2qfp_A |
424 |
269 |
135 |
2.49 |
16.30 |
32.433 |
5.222 |
T P |
| 2hy1_A |
227 |
269 |
131 |
2.38 |
16.03 |
31.815 |
5.292 |
T P |
| 2a22_B |
203 |
269 |
124 |
2.49 |
12.90 |
29.408 |
4.782 |
T P |
| 2qjc_A |
222 |
269 |
117 |
2.44 |
14.53 |
28.564 |
4.603 |
T P |
| 1xm7_A |
186 |
269 |
94 |
2.47 |
10.64 |
23.058 |
3.659 |
T P |
| 2pb1_A |
394 |
269 |
93 |
2.77 |
8.60 |
21.655 |
3.245 |
T P |
| 3e70_C |
307 |
269 |
74 |
2.57 |
6.76 |
18.986 |
2.771 |
T P |
| 2lbp_A |
346 |
269 |
77 |
2.67 |
9.09 |
18.794 |
2.779 |
T P |
| 3dm5_A |
416 |
269 |
74 |
2.46 |
2.70 |
18.574 |
2.887 |
T P |
| 2dwu_B |
267 |
269 |
74 |
2.49 |
5.41 |
18.475 |
2.852 |
T P |
| 1usg_A |
345 |
269 |
73 |
2.71 |
10.96 |
16.191 |
2.594 |
T P |
| 2jfp_A |
268 |
269 |
63 |
2.46 |
9.52 |
15.926 |
2.461 |
T P |
| 2vvt_A |
270 |
269 |
59 |
2.62 |
6.78 |
14.500 |
2.168 |
T P |
| 3cvu_A |
501 |
269 |
55 |
2.72 |
9.09 |
12.854 |
1.948 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]