LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_98.5wLII_11068_57
Total number of 3D structures: 34
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
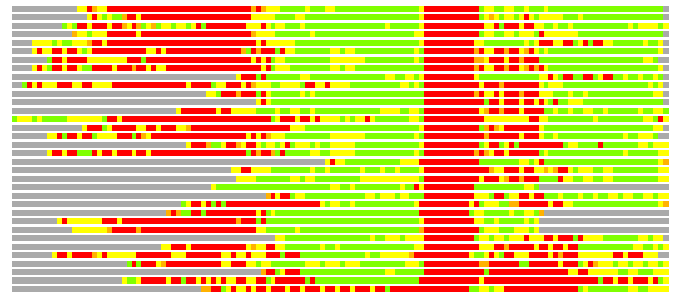
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1qu7_A |
227 |
132 |
76 |
1.87 |
10.53 |
51.612 |
3.861 |
T P |
| 2ch7_A |
309 |
132 |
79 |
2.06 |
8.86 |
50.787 |
3.654 |
T P |
| 1cii_A |
602 |
132 |
86 |
2.16 |
6.98 |
50.724 |
3.806 |
T P |
| 2ch7_B |
307 |
132 |
79 |
2.05 |
7.59 |
50.453 |
3.670 |
T P |
| 1deq_A |
180 |
132 |
76 |
2.10 |
9.21 |
47.161 |
3.450 |
T P |
| 2dw4_A |
634 |
132 |
74 |
2.14 |
5.41 |
46.697 |
3.303 |
T P |
| 2iw5_A |
666 |
132 |
73 |
2.20 |
5.48 |
46.046 |
3.178 |
T P |
| 2z3y_A |
643 |
132 |
75 |
2.20 |
1.33 |
45.889 |
3.255 |
T P |
| 1wyy_A |
126 |
132 |
65 |
1.77 |
9.23 |
44.825 |
3.481 |
T P |
| 2hko_A |
647 |
132 |
75 |
2.34 |
5.33 |
44.637 |
3.075 |
T P |
| 1jch_A |
468 |
132 |
67 |
2.04 |
4.48 |
42.982 |
3.131 |
T P |
| 2dq3_A |
425 |
132 |
60 |
1.64 |
5.00 |
42.763 |
3.457 |
T P |
| 1y1u_A |
544 |
132 |
70 |
2.10 |
8.57 |
42.229 |
3.176 |
T P |
| 1f5n_A |
570 |
132 |
78 |
2.27 |
5.13 |
41.527 |
3.284 |
T P |
| 2b5u_A |
470 |
132 |
65 |
2.08 |
6.15 |
41.232 |
2.981 |
T P |
| 2v1d_A |
666 |
132 |
72 |
2.17 |
4.17 |
41.171 |
3.174 |
T P |
| 1qvr_A |
803 |
132 |
65 |
2.19 |
4.62 |
40.752 |
2.840 |
T P |
| 2h94_A |
647 |
132 |
71 |
2.13 |
5.63 |
40.357 |
3.181 |
T P |
| 2wb7_A |
525 |
132 |
55 |
1.73 |
5.45 |
39.072 |
3.010 |
T P |
| 3cwg_B |
507 |
132 |
66 |
2.36 |
4.55 |
38.834 |
2.687 |
T P |
| 1bg1_A |
559 |
132 |
67 |
2.24 |
4.48 |
38.474 |
2.863 |
T P |
| 1m1j_C |
390 |
132 |
55 |
1.55 |
1.82 |
38.291 |
3.336 |
T P |
| 2dnx_A |
130 |
132 |
62 |
2.22 |
6.45 |
38.125 |
2.674 |
T P |
| 1lrz_A |
400 |
132 |
56 |
2.11 |
3.57 |
36.993 |
2.533 |
T P |
| 1ei3_C |
397 |
132 |
52 |
1.81 |
3.85 |
36.776 |
2.728 |
T P |
| 2efr_A |
155 |
132 |
56 |
1.89 |
10.71 |
36.408 |
2.810 |
T P |
| 1c1g_A |
284 |
132 |
55 |
1.96 |
3.64 |
35.910 |
2.663 |
T P |
| 1bf5_A |
545 |
132 |
61 |
2.09 |
6.56 |
35.749 |
2.785 |
T P |
| 2r05_A |
697 |
132 |
62 |
2.13 |
11.29 |
35.465 |
2.777 |
T P |
| 3fhn_A |
673 |
132 |
66 |
2.77 |
6.06 |
34.943 |
2.297 |
T P |
| 2oev_A |
697 |
132 |
64 |
2.46 |
3.12 |
34.692 |
2.503 |
T P |
| 2jqq_A |
154 |
132 |
47 |
2.03 |
4.26 |
31.087 |
2.210 |
T P |
| 1u0b_B |
461 |
132 |
44 |
2.19 |
6.82 |
26.803 |
1.924 |
T P |
| 2iub_F |
342 |
132 |
40 |
2.54 |
5.00 |
22.842 |
1.515 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]